
Article highlights
- Chronic wounds are a major socioeconomical healthcare burden
- Identification of beneficial therapeutics hindered by the lack of an ideal chronic wound animal model
- None of the existing animal models of delayed healing fully recapitulate human chronic wounds
- Chronic wounds are multifactorial in nature and arise through a combination of factors
- Delayed models of healing, including the streptozotocin diabetic model, skin flap model and magnet-induced IR models have emerged
- An ideal chronic wound model should include features such as delayed re-epithelialization, hyper thickened non-migratory wound edges with overexpression of the gap junction protein connexin 43, persistent inflammation, elevated ROS levels, alkaline wound environment, excessive extracellular matrix (ECM) degradation at wound edges, disrupted/impaired vasculature and sustained presence of senescent cells
This box summarizes key points contained in the article.
1. Introduction
Numerous therapeutics and medical devices fail in clinical trials despite success in animal models. The lack of advances in chronic wound care can be largely attributed to the absence of animal models that recapitulate features of human chronic wounds. Animal models are crucial to understanding the underlying cellular and molecular mechanisms that hinder wound healing. Greater knowledge of these mechanisms could expedite the identification of potential therapeutic targets to trigger or even accelerate chronic wound closure. The complexity of human chronic wounds hinders the development of animal models as they lack many of the features seen in man.
Chronic wounds typically develop from a combination of different etiological factors[1,2]. Healing outcomes of problematic wounds can be adversely affected by patient comorbidities such as venous insufficiency and diabetes[3]. Chronic wounds rarely develop in animals due to differences in skin architecture and wound healing mechanism[4,5]. In animals, only the thoroughbred racehorse develops chronic wounds[6]. Deliberate intervention in the healing process would be required to develop delayed wound healing animal models.
Currently, most animal models of delayed healing focus on isolating the primary clinical causes of chronic wounds – diabetes, ischemia, and reperfusion damage[7,8]. Various factors should be considered when determining the most appropriate animal model for testing wound healing therapeutics. Some factors include reproducibility, physiological relevance to human chronic wounds, tractability, and cost effectiveness. In this review, we will highlight commonly used animal models of impaired healing. The advantages and limitations of each model will be discussed. To evaluate the clinical relevance of each model, histological features such as delayed wound closure and wound edge hyper thickening will be compared with the key features of human chronic wounds.
2. Chronic wounds and their socioeconomic burden
Chronic wounds fail to restore function and tissue integrity though the normal reparative process. These wounds can persist for months or even years[7]. Of note, there are many different types of chronic wounds. However, chronic wounds arising from surgical and infectious wounds remain rare. Here, we focus on the three main types – diabetic foot ulcers,pressure ulcers, and venous leg ulcers[8]. Chronic wounds inflict a huge socioeconomic burden, affecting nearly 2.5% of the total population in the United States [9]. The management of chronic wounds has been an enormous drain on healthcare resources and in the United States of America (USA) it is estimated to cost an excess of US$28.1 billion annually[10]. Aside from financial implications, patients also suffer a significant reduction in quality of life[11]. With an increase in ageing populations and incidence of lifestyle diseases such as obesity, the prevalence of chronic wounds will continue to rise[12].
3. Comparison between acute and chronic wounds
The tissue repair process involves complex interactions between multiple cell types and the extracellular matrix. Wound healing follows a precisely orchestrated series of events. The events are coordinated by soluble mediators such as growth factors and cytokines. The process is divided into four distinct but overlapping phases – hemostasis, inflammation, proliferation, and tissue remodeling[13,14]. A more detailed summary of the wound healing events is illustrated in Figure 1. In contrast, a chronic wound fails to adhere to these sequences of events and get stuck in the proinflammatory phase. Local factors such as ischemia, tissue maceration and infection can have adverse effects on the normal reparative process. Systemic factors such as age, malnutrition, elevated glucose and comorbidities such as vascular insufficiency and diabetes can further complicate the wound healing process[1]. Reduction in migrative capabilities, chronic inflammation and elevation of proteolytic enzymes and reduction of their inhibitors are also hallmarks of chronic wounds. The following section explores and further discusses the various characteristics of chronic wounds.
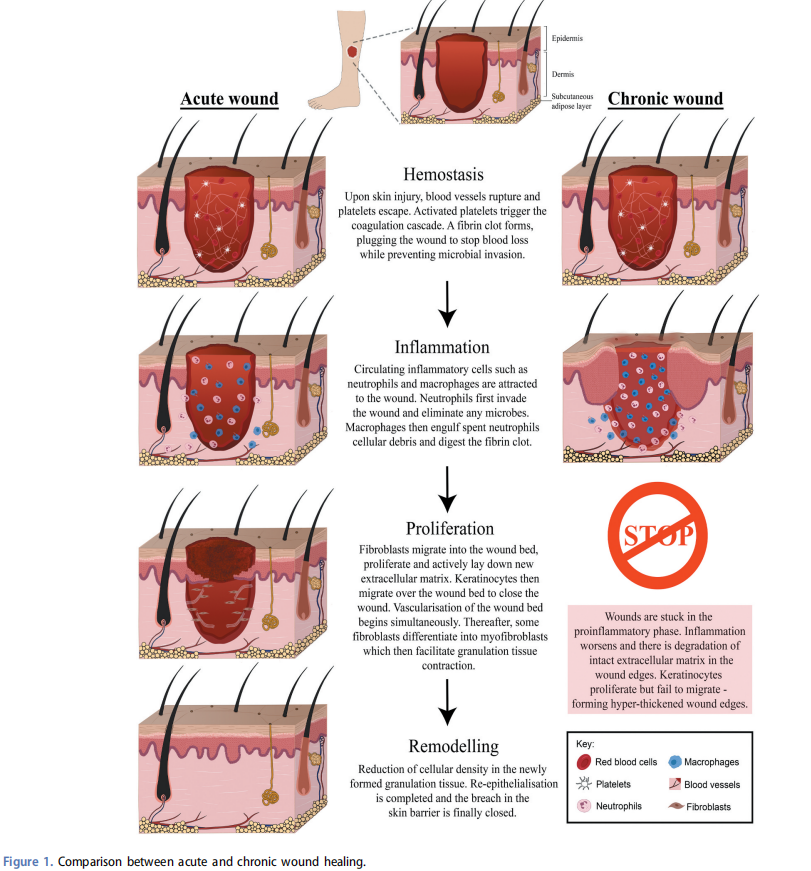
3.1. Stalled re-epithelialization and hyper thickened wound edges
Re-epithelialization is a critical process for restoration of the epidermal barrier[15]. To initiate epithelial repair, wound edge keratinocytes change from their natural homeostatic behavior and become migratory[16]. Before migrating, keratinocytes become activated and break down their cell-cell and cellmatrix adhesion[17]. Subsequently, active migration at the wound border occurs through the formation of a thin migratory tongue which crawls over a provisional granulation tissue matrix [15]. Behind these migratory cells, a cluster of highly proliferative cells multiply to feed in new cells to sustain the migration[18].
In chronic wounds, re-epithelialization fails to occur[19]. This delay can be attributed to multiple factors such as tissue hypoxia, excessive pro-inflammatory cytokines and bacterial infection. Keratinocytes at chronic ulcer margins frequently express genes associated with keratinocyte activation and hyper-proliferation –Keratin 16 (K16) and Ki67 respectively[20]. However, genes associated with keratinocyte differentiation and migration are minimally expressed – K10 and K2 respectively[21]. Consequently, the wound edge forms a piled-up and hyper proliferative epidermis that exhibits a non-migratory phenotype[20].
3.2. Chronic inflammation
Following skin injury, blood vessels rupture. Platelets are released and activated, triggering a cascade of reactions that ultimately leads to the formation of a fibrin clot[14]. Activated platelets release growth factors and cytokines such as platelet derived growth factor, which act as chemoattractants to inflammatory cells[14]. Neutrophils dominate the early phase of inflammation, releasing reactive oxygen species (ROS) and proteases to eliminate bacteria[22]. Subsequently, phagocytic macrophages clear spent neutrophils and scavenge remaining tissue debris[22]. The acute inflammatory response is selflimiting and requires complete resolution to restore tissue homeostasis. In contrast, chronic wounds are stuck in a selfperpetuating state of inflammation[23].
Adequate clearance of apoptotic cells from wound sites is a pre-requisite for successful restoration of normal tissue function[24]. Chronic wounds often display a build-up of dead cell debris at wound margins[7]. This can be attributed to decreased phagocytic ability of immune cells[7]. Impairment to efferocytosis exposes the wound site to toxic contents of dying cells[24]. Concurrently, increased apoptotic cell burden in the wound bed leads to elevated levels of pro-inflammatory cytokines[25].
Inflammation is regulated by pro- and anti-inflammatory signals that initiate, maintain, and resolve inflammation [24]. Imbalance in these signals, in favor of a proinflammatory state, derails the healing cascade[26]. Proinflammatory cytokines, such as tumor-necrosis factor alpha (TNF-α) and interleukin-1beta (IL-1β), are prominently upregulated [27]. Perturbation of the delicate equilibrium between inflammatory signals results in the perpetuation of tissue damage[26].
3.3. Imbalance between proteolytic enzymes and their inhibitors
Degradation and remodeling of the extracellular matrix (ECM) drives cellular migration, granulation formation and angiogenesis[28]. In particular, matrix metalloproteases (MMPs) are the key enzymes involved in cleavage of ECM proteins[28]. During different phases of wound healing, MMPs are secreted by inflammatory cells, fibroblasts, keratinocytes, and endothelial cells[29]. These enzymes are essential in every phase of wound healing as controlled proteolytic degradation is critical for successful tissue repair[29]. This process is tightly regulated by the tissue inhibitors of MMPs (TIMPs)[28].
In contrast, sustained proteolytic activity due to elevated MMP levels is one of the prime underlying factors for abnormal healing in chronic ulcers[30]. MMPs, such as MMP-2 and MMP-9, and serine proteases are frequently found at significantly higher levels in chronic wound fluids[30–32]. This increase in proteinase level can be attributed to uncontrolled levels of pro-inflammatory cytokines – a prominent characteristic of chronic ulcers[1]. Furthermore, levels of TIMPs that are crucial in inhibiting proteolytic activity are also decreased in chronic wounds[33]. Consequently, an imbalance between MMP and TIMPs leads to excessive degradation of the native ECM[28]. Additionally, the synthesis of new ECM proteins is also disrupted[28]. Ultimately, the inability to provide a functional ECM hinders the healing process.
3.4. Sustained presence of senescent cells
The sustained presence of elevated levels of senescent cells in the wound is detrimental to healing[34]. Various triggers such as deoxyribonucleic acid (DNA) and oxidative damage, as well as mechanical stress can induce cellular senescence[35]. Consequently, cells are driven into a state of cell cycle arrest. Nonetheless, senescent cells can play beneficial roles in wound healing through the secretion of growth factors and ECM proteins[36]. Specifically, platelet-derived growth factorAA (PDGF-AA) has been shown to promote fibroblast differentiation, proliferation, and granulation tissue formation[36]. However, timely clearance of senescent cells by macrophages is critical[37]. Transient induction of senescence promotes tissue repair. In particular, the levels of senescent cells peaked 6 days following wounding, and then returned to basal levels 9–12 days post-wounding[36]. Conversely, persistent senescence can lead to disrupted wound healing processes[34]. In particular, accumulation of senescent cells is commonly observed in chronic wounds. Senescent cells express a hypersecretory phenotype known as senescence-associated secretory phenotype (SASP)[38]. Negative wound healing outcomes have been closely associated to SASP factors[37]. These factors include proteases and pro-inflammatory cytokines that shift the wound environment into a highly inflamed and proteolytic state – degrading native ECM and hindering cell migration over newly formed matrices in chronic wounds[38]. Moreover, sustained SASP expression can also lead to excessive inflammatory cell recruitment and retention[37]. Eventually, this exacerbates the heightened inflammatory state present in chronic wounds[37]. Taken together, chronic senescence impedes tissue repair[37].
3.5. Low oxygen levels
Upon injury, oxygen levels decrease, stimulating the early phase of wound healing[39]. Transient hypoxia triggers the production of ROS, cytokines and growth factors[40]. These molecules coordinate immune cell recruitment, promote vascularization, and encourage cell proliferation and migration [39]. However, recovery of wound tissue oxygenation is essential to drive the later phases of the wound healing process[41]. Typically, oxygen levels are restored through the formation of new vasculature from areas adjacent to the wound, in a process called angiogenesis[14].
While acute hypoxia is beneficial, prolonged oxygen deprivation impairs healing[42]. Patients with diabetes, arterial or venous diseases commonly suffer from vascular complications that leads to chronic hypoxia[42]. Furthermore, angiogenesis is impaired in chronic wounds[43]. Angiogenic factors such as vascular endothelial growth factor (VEGF) and VEGF-receptor-1 (VEGFR-1) are upregulated in chronic ulcers[44]. However, the highly proteolytic chronic wound environment destroys VEGFR-1 and renders these angiogenic factors ineffective in supporting vascularization[44]. Persistent hypoxia impairs the wound healing cascade and eventually disrupts tissue repair[42].
Overproduction of ROS increases oxygen demand and further contributes to the hypoxic chronic wound environment[45]. Chronic wounds suffer from a proinflammatory environment that encourages and sustains the presence of inflammatory cells such as macrophages and neutrophils[26]. Consequently, there is an overproduction of ROS[42]. Hence, cells in the chronic wound environment suffer from oxygen deprivation[42]. Furthermore, excessive ROS perpetuates inflammation, activates proteolytic enzymes and further provokes tissue damage in chronic wounds[46].
3.6. Chronic infection with bacterial biofilms
Open wounds provide opportunities for microbes to infect the wound site. In response, the host immune system mounts a defense mechanism to eliminate bacteria[4]. Ischemic and necrotic tissues in ulcers increase susceptibility of the wound to bacterial infection[47]. These bacteria form biofilms and are prevalent in 60% of chronic wounds. In contrast, only 6% of acute wounds contain biofilms[48]. Bacteria present within the biofilms often demonstrate increased resistance towards immune attack and antimicrobials[49]. In particular, virulence factors such as rhamnolipids, secreted by Pseudomonas aeruginosa (P.aeruginosa), confer protection against phagocytosis [50]. Consequently, elevated levels of microbial components stimulate production of pro-inflammatory cytokines through toll-like receptor (TLR) activation pathways[50]. Combined with an impaired host response, persistent bacterial load eventually contributes to the prolonged inflammatory phase of chronic wounds.
4. Animal models of delayed wound healing
The complexity of human chronic wounds hinders the development of clinically relevant animal models. So far, no single animal model is capable of fully recapitulating the different features of human chronic wounds, described above. However, existing models are still useful therapeutic platforms that reflect different elements of human chronic ulcers. This includes but is not limited to delayed wound models for surgical, cancerous, diabetic and ischemic wound conditions. Here, we focus on the most commonly used delayed wound models and describe the induction and relevance of these models in hopes of further understanding chronic wound pathology. Concurrently, the limitations of each model are also discussed to aid in the refinement and development of new models. A summary of these different models of delayed healing and their observed histological features, in relation to chronic wounds, is highlighted in Table 1.
4.1. Ischemic wounds
Localized tissue ischemia is a key underlying feature of chronic wounds[4]. In particular, vasculature is commonly destroyed or has abnormalities that reduce blood flow – limiting oxygen supply and preventing metabolic waste removal[4]. Oxygen deprivation can lead to the impairment of mitochondrial oxidative phosphorylation, which eventually triggers overproduction of ROS[51]. Hence, oxygen deficit in a chronic wound is exacerbated. Moreover, the accumulation of metabolic waste coupled with excessive ROS in the wound bed further stimulate undesired inflammatory responses and cause additional tissue damage[52].
Several ischemic wound models have been developed over the years. Most of these models are based on the deprivation of blood supply to a specific area to induce an ischemic region. Utilization of these clinically relevant models have been useful in investigating the contribution of ischemia to delayed wound healing[24,53,54]. In addition, the efficacy of topical growth factors, such as PDGF-β[55], and VEGF[56] treatments have been tested on these ischemia-induced wounds.
4.1.1. Methods of induction
4.1.1.1. Ischemic rabbit ear model.
One of the most extensively used ischemic models involves the creation of a cutaneous ischemic zone through arterial ligation of a rabbit’s ear[57,58]. A circumferential incision is made at the base of the ear. Thereafter, two of the three arteries (central and caudal) are ligated while leaving the veins and cartilages intact. The resulting disruption of blood flow and consequently oxygen supply creates an ischemic environment. A wound is then created using a 6 mm biopsy punch down to the cartilage. Due to firm attachment of the cartilage to the underlying dermis, the avascular wound bed does not close by contraction[57].
Wound healing is significantly impaired in this model. These ischemic wounds failed to close even at 7 days postwounding[57,58]. In contrast, control wounds showed extensive re-epithelialization and granulation tissue formation [57,58]. Although there are some signs of epithelial tongue migration at 10 days post-wounding, the granulation tissue in these ischemic wounds remained notably deficient[57,58].
4.1.1.2. Skin flap surgery.
Another model to study ischemia is the skin flap model. Flaps are first made on the back of animals to partially detach specific regions of the skin, thereby disrupting circulatory flow to the region and creating a localized hypoxic zone[53]. This zone of hypoxia is sustained by placing a sheet of material (e.g. silicone) beneath the flap to prevent its adherence while limiting perfusion and revascularization into this region[59]. Full-thickness wounds, made with 6 mm biopsy punches, are then created in these skin flaps to generate ischemic wounds.
On average, ischemic wounds, generated from this model, healed in 14–21 days [53,60]. In contrast, control wounds demonstrated complete closure after 10–12 days[53,60]. Moreover, pro-inflammatory cytokines and protease activity was elevated in these ischemic wounds when induced on both rats and pigs[53,54].
4.1.2. Limitations of ischemic wound models
While both models mentioned in the previous sub-sections can induce ischemic wounds, there are several limitations that should be considered when evaluating their relevance to human chronic wounds. While the ischemic rabbit ear model could potentially be induced on rodents, the size of rodents’ ears limits the feasibility and reproducibility of this model[59]. Moreover, the induction of this model requires extensive anatomical knowledge and fine surgical skills. On the other hand, the skin flap model is a more accessible ischemic wound model to researchers. However, the variability in extent of ischemia and the corresponding tissue damage is a limitation. In particular, irregular vascular distributions within each skin flap and between the skin flaps on large animals contribute to poor reproducibility of ischemic wound healing outcomes[61].
Beyond the above-mentioned factors, the ischemic environment induced by both models is transient. New vessels develop after 10–14 days in the ischemic rabbit ear model [62] while blood supply returns to normal levels 2–4 weeks in the skin flap model[63,64]. In contrast, human chronic wounds frequently suffer chronic and persistent blood insufficiency[1]. Furthermore, unlike the gradual reduction in perfusion pressure in human chronic ulcers, both models induce ischemia in an abrupt fashion [65,66].
4.2. Ischemia-Reperfusion injuries
The etiology of ischemia-reperfusion (IR) injury is distinct from that of a single ischemic insult. IR injury refers to the secondary tissue damage when blood returns to a tissue following a period of ischemia[8]. Although re-establishment of blood flow is essential to reversing ischemia, the reperfusion process paradoxically causes further damage. In particular, the production of excessive oxygen free radicals exacerbates the local inflammatory response[4].
Pressure ulcers can form from unrelieved mechanical pressure over susceptible areas, such as bony prominences[67]. Notably, the aged and immobile are most prone to developing pressure ulcers[68]. Clinically, patients with limited mobility are re-positioned regularly to relieve pressures on bony prominences[69]. However, in recent years, reperfusion has emerged as an important contributing factor towards the development and progression of pressure ulcers[8].
4.2.1. Methods of induction
Several IR-based approaches have been employed to induce pressure ulcer injury in animals. These models have been useful towards the investigation of the underlying mechanism of different stages of pressure ulcer formation as well as in the development of new therapeutic interventions. In particular, antioxidants such as melatonin[70] and the effects of compounds such as heparin sulfate glycosaminoglycan mimetic [71] have been tested in these models.
These approaches are centered on subjecting the skin to multiple cycles of ischemia and reperfusion[59]. Specifically, cyclic compression can be achieved through two commonly utilized methodologies: the implanted model and the pinch model. Briefly, the implanted model involves the surgical insertion of a magnet or steel plate beneath the skin[72]. Subsequently, an external magnet is applied, creating an ischemic region [72]. Alternatively, the pinch model creates localized ischemia through the application of two magnets on either side of tented skin[73]. In comparison to the implanted model, this technique eliminates the use of invasive procedures and generates two areas of IR insult. The removal of the magnet allows reperfusion injury to occur. Through repeated application and removal of the magnet, ischemia-reperfusion injury is induced. Parameters such as magnet strength as well as IR cycle frequency and duration can be altered to induce IR injuries of varying severity[72,73]. Alternatively, other methods of blocking the blood supply can also be achieved using clamps and lead weights. In addition, skin flap models have been used in conjunction with clamping in the study of flap survival and the effects of ischemic pre-conditioning. Briefly, an island flap is created, and a silicon sheet barrier is placed following elevation of the skin flap. Subsequently, ischemicpreconditioning is carried through the use of microclamps to undergo multiple ischemic-reperfusion cycles[74].
IR injuries induced in animals using either method have presented features comparable to human pressure ulcers[75]. In particular, increased leukocyte infiltration, non-blanched skin, vascular leakage, and epidermal thickening were observed in less severe IR models[73]. These features mimic the early stages of pressure ulcers[75]. Varying the parameters such as the frequency of IR cycles allows the induction of more severe IR injuries that mimic the later stages of human pressure ulcers. Notably, open wounds with delayed closure were observed[76]. These wounds also had increased protease levels (e.g. MMP-9 and MMP-13) that further contributed to excessive collagen loss in the dermis[76].
4.2.2. Limitations of ischemic-reperfusion model
There are two notable limitations arising from the implanted model when compared to the pinch model: the invasive surgical procedure and foreign body reaction. Infections could develop at surgical incision sites and complicate the injury outcomes. Moreover, the implantation of the steel plate or magnet is known to induce a foreign body reaction[73]. The resultant inflammatory response cannot be entirely attributed to the IR injury.
Overall, there has been a lack of consensus on the different parameters (magnet type, magnet strength, number and duration of IR cycles) employed in inducing IR injury. The comparison of results across different studies remains challenging. Importantly, there is no animal with a loose skin architecture similar to that of aged skin in humans. So far, most of the IR injury models are conducted in rodents due to their loose skin architecture[77]. However, rodents have an additional panniculus carnosus layer that is absent from human skin[59]. So the relevance of these IR models to human pressure ulcers is limited.
4.3. Diabetic wounds
Diabetes can be divided into two major classifications – Type 1 and Type 2 diabetes[78]. In Type 1 diabetes, autoimmune destruction of the beta cells causes insulin deficiency[78]. Conversely, insulin resistance is a critical factor in the development of Type 2 diabetes[79]. Moreover, the beta cells are unable to compensate by increasing insulin secretion[78]. Hence, a combination of these factors contributes to Type 2 diabetes progression.
Diabetic patients suffer from hyperglycaemia[80]. Sustained high blood glucose levels causes elevated ROS levels, chronic inflammation, DNA damage and induction of senescence – all of which impair wound healing[80]. In addition, non-healing ulcers often become infected increasing the risk of sepsis and necrosis. Collectively, this can eventually lead to limb amputation[81]. In Singapore alone, at least four diabetes-related lower leg amputations occur daily[82].
4.3.1. Methods of induction
Diabetes can be induced in animals through spontaneous mutations as well as genetic or chemical manipulation[7,79]. There are many diabetic mouse strains however, the two most common genetic strains used to study diabetic wound healing are Akita and db/db mice[7]. Conversely, genetically modified diabetic strains such as NONcNZ0101/LtJ mice are also frequently used. Other mouse models that have been used to study diabetes also include the NOD, ob/ob and NZO mice[79]. Although genetically modified diabetic pigs have also been developed for diabetic research, to the best of our knowledge, these have not yet been adopted for wound healing studies [83]. Alternatively, the introduction of chemical agents such as streptozotocin (STZ) or alloxan can induce diabetes in both rodents and pigs[79].
The development of different diabetic animal models has provided valuable insights into the progression of diabetes and chronic wounds[84]. In addition, these models have been useful in testing the efficacy of a wide range of therapeutics such as growth factors[85] and pro-angiogenic drugs[86]. However, no single model can completely reflect the pathogenesis of both Type 1 and Type 2 diabetes. Ideally, the choice of animal model will depend on its relevance to a certain aspect of each disease.
4.3.1.1. Genetically induced diabetic models
4.3.1.1.1. Akita mice model.
Akita mice develop a type 1 diabetes phenotype through an autosomal dominant missense mutation of the insulin 2 (Ins2) gene which causes abnormal folding of the insulin protein[78]. Consequently, this causes toxic injury to pancreatic beta cells and eventually decreases insulin secretion[87]. After 4 weeks from birth, these mice develop features such as insulinopenia and hyperglycemia[78]. In particular, homozygous akita mice can suffer from a premature death due to the development of more severe phenotypes than their heterozygous counterparts[87]. In addition, male mice also develop a more severe hyperglycemic phenotype and may require insulin administration to survive [78]. 6 mm splinted wounds induced on akita mice show delayed healing. In particular, these wounds show statistically significant delay in wound closure compared to wounds on wild-type mice – 17 days and 12 days respectively[88].
4.3.1.1.2. db/db mice model
Leptin is a hormone that mediates appetite[79]. The binding of leptin to its receptor suppresses hunger[79]. Here, the genetic basis of the db/db model involves the inactivation of the leptin receptor, leading to defective leptin signaling and the inability to simulate satiety. Consequently, food intake increases and eventually these mice become obese[79]. These mice also progressively develop other diabetic symptoms such insulin resistance and hyperglycemia[89], similar to the progression of Type 2 diabetes in humans. At 4–8 weeks of age, db/db mice are found to have significantly elevated blood sugar levels[89].
There are many delayed wound healing features present in wounds induced on the db/db model. However, many of these features are induced in a splinted db/db wound model so as to minimize the wound contraction largely contributed by the panniculus carnosus[90]. These features include delayed wound closure, enhanced inflammatory response, increased senescent cell populations[91], reduced granulation tissue formation, and decreased vascularity[88,92]. In particular, db/db mice were observed to have slower wound closure rates than wild-type mice with a 6 mm diameter wound – 20 to 22 days and 11 to 13 days respectively[88,90].
4.3.1.1.2. NONcNZ010/LtJ mice model.
The NONcNZ010 mice represent a new polygenic strain developed to resemble obesity-induced Type 2 diabetes more closely[93]. Specifically, this model is a recombinant congenic strain generated through the combination of quantitative trait loci from two mice strains, the NZO/HILT and NON/LtJ[93]. The resultant mice develop insulin resistance and hyperglycemia[93]. In addition, this diabetes phenotype is independent of diet and is not characterized by an overtly dysregulated leptin gene expression[94]. Consequently, the NONcNZ010 strain produces a moderate form of obesity instead of the massive obesity seen in leptin-related models[94]. 6 mm diameter wounds on NONcNZO010 mice have significantly impaired wound healing rates. Mean wound healing rates for wounds on wild-type mice were 0.208 mm/day compared to 0.076 mm/day in wounds on NONcNZO010 mice[95].
4.3.1.2. Chemical induction of diabetes.
Streptozotocin and alloxan are the two most commonly used chemical compounds for Type 1 diabetes induction in animals[7]. These compounds are glucose analogs that affect the glucose transporter 2 (GLUT2) of beta cells[96]. Subsequently, beta cells within the islets are completely ablated, compromising insulin production and eventually leading to hyperglycemia and weight loss[96]. Of note, although both chemical agents can induce diabetes in animals, STZ is more toxic to rabbits and hence, alloxan is adopted as a substitute for rabbit work[97].
To date, the STZ model is one of the most commonly adopted diabetic models to study delayed wound healing [79]. Accordingly, mild to severe diabetes can be produced by varying parameters such as dosage, strain, age, and route of administration. For instance, a single STZ dose injected into rats can range from 35 mg/kg to 60 mg/kg[98]. After 3 weeks, these rats develop diabetes with blood glucose levels falling between 250 and 600 mg/dL (13.9 to 33.3 mmol/L)[98]. A similar approach can be adopted in pigs[78].
In a splinted STZ diabetic rat model, delays in wound healing were observed[88]. Specifically, 6 mm diameter wounds on STZ rats closed at day 15 compared to nondiabetic controls which showed complete wound closure at 12 days post wounding[88]. In STZ rat wounds, other features such as hyper thickened wound edges, lack of blood vessels in the granulation tissue, increased inflammation, increased MMP-9 levels and decreased TIMP-1 expressions are also observed[99,100].
Similarly, STZ induced diabetic pigs show significantly delayed wound re-epithelialization compared to non-diabetic pigs. Specifically, for a 1.5 cm by 1.5 cm full-thickness wound, closure was observed at after 12–14 days for non-diabetic pigs compared to 18 days for diabetic pigs[101].
4.3.2. Limitations of diabetic wound models
Currently, none of these models can completely recapitulate the variations and different pathological processes of human diabetic wounds[59]. Each of these models only serve to represent a limited aspect of this complex disease.
Most of the diabetic mice strains have a monogenic inheritance pattern. In contrast, human type 2 diabetes is multifactorial and polygenic in nature[94]. In particular, the key feature of leptin-related diabetic models is the development of severe, early-onset obesity[88]. However, obesityrelated type 2 diabetes in humans are rarely caused by a monogenic mutation and can occur at any point throughout life[94]. Moreover, fasting blood glucose plasma levels in db/db mice can reach up to 600 mg/dL[102]. This level of hyperglycemia is extreme compared to human type 2 diabetes, which typically exhibit an average fasting plasma glucose (FPG) level of 200 mg/dL[103].
Diabetic mice strains and genetically modified rodent models have been predominantly used in diabetes research. However, there are limitations due to differences with human skin anatomy and wound healing mechanisms[59]. Advances in genetic engineering have facilitated the generation of diabetes in large animals, such as the pig, which more closely mimics human disease[83]. In particular, transgenic pigs expressing modeling maturity onset diabetes of the young 3 (MODY 3) have been generated. These pigs express a dominant-negative hepatocyte nuclear factor-1 alpha (HNF1A) that leads impaired insulin secretion and eventually, the development of persistent diabetes[83]. Despite the availability of genetically induced diabetic pigs, so far, none of them have been used for wound healing studies.
Although chemically induced diabetic models have advantages such as lower costs and simpler induction methods, they also have their limitations. Notably, there is no standardized consensus on the parameters for STZ induction. This is partly caused by the difference in sensitivity to STZ in various rodent strains and genders[104]. Additionally, the length of time between diabetes induction and subsequent wounding also influences wound healing outcomes [100]. A trend towards delayed re-epithelialization was only observed 2–3 weeks post STZ induction in rats[99,100]. In addition, impairment of angiogenesis and collagen deposition required longer timelines up to 6 weeks post diabetic induction[100]. However, earlier timepoints of less than 3 weeks are frequently used in many studies[100]. Limitations in STZ-induced diabetes in larger animals such as pigs are often associated with reduced responsiveness towards STZ due to low GLUT2 levels[105]. Although increasing STZ dosages can compensate for the low STZ sensitivity in pigs, however, other complications such as hepatic and renal toxicity can arise[78].
4.4. Biofilm-infected wounds
Despite the association of biofilms with negative wound healing outcomes, the significance and mechanism of biofilms in chronic wounds remains poorly understood. In particular, the dynamic interactions between different bacterial species and their resultant effect on delayed wound healing represents a void in our understanding. Many wound infection models have been used to study the interactions of different bacteria, commonly found in chronic wounds, and the resultant effect on wound healing[106]. In addition, these models have been used to investigate the therapeutic efficacy of antimicrobial products[107] and various wound care products on bacterial biofilms.
4.4.1. Methods of induction
Wound samples from chronic ulcer support mixed species of bacteria. However, P. aeruginosa and Staphylococcus aureus (S. aureus) are the most commonly used to generate biofilminfected wound models (as they are the most common bacteria to be found in chronic wounds). Typically, bacterial cells can be applied as planktonic cells (104 –106 colony forming units/wound) to wounds[59]. Alternatively, bacteria can also be delivered via a bacterial contaminated implanted material or pre-formed biofilm on filter paper[108–110]. Thereafter, occlusive dressings are placed over infected wounds to prevent cross-contamination and provide optimal bacterial growth conditions. 6 mm diameter wounds in rodents infected with biofilms reached closure between 18–21 days [110]. In comparison, non-infected wounds reached closure between 9–12 days[110]. Additionally, the presence of biofilm induced an increased and sustained inflammatory response as well as decreased formation of granulation tissue[111].
4.4.2. Limitations of biofilm-infected models
Unlike chronic wounds, which are polymicrobial in nature,most established biofilm-infected models are based on the use of a single dominant wound microbial species[112]. Moreover, infections in these models are typically short-term ranging between 2 and 26 days[106]. In contrast, clinically, prolonged host-biofilm interactions are known to complicate the wound microenvironment[113]. Hence, these biofilminfected models are unable to recapitulate the complexity and persistent nature of biofilms in chronic wounds[48]. In addition, motile bacteria such as P. aeruginosa can move between wounds and thus complicate the use of controls in the same animal. Furthermore, the application of bacteria to wounds is relatively well-tolerated in young healthy animals. Consequently, bacterial infections at wounds can be easily overcome and cleared away[114]. Although application of higher bacterial loads can increase the success of generating biofilm-infected models, the animal could suffer from systemic infection and die[114].
4.5. Other possible approaches to induce delayed wound healing
The models discussed above primarily focus on isolating the main causative factors of chronic wounds – local tissue hypoxia, biofilm colonization, repetitive ischemia-reperfusion injury, and diabetes. However, chronic wounds are multifactorial[1,2]. Although each of these conditions can be deleterious, collectively they can overwhelm the normal healing response. Hence, many studies couple two or more delayed wound models to generate more clinically relevant models[7]. One of the most frequently used approach is the application of biofilms on a db/db diabetic mouse[115]. In this model, even in the absence of using a splint to counter wound contraction, all of the biofilm-infected wounds remained open at 28 days post-wounding[115]. This is a stark contrast when compared to splinted wound models on db/db mice that closed at 20–22 days post wounding[88]. In addition to the delayed wound closure, epidermal hyperplasia was also observed these biofilm-infected diabetic wounds, which is a histological feature clearly absent in uninfected wounds on db/db mice[115].
5. Expert opinion
Approximately 89% of novel drug discoveries fail to pass human clinical trials, due to failure of efficacy or unanticipated human toxicity[116]. This raises important concerns regarding the reliability and predictive value of animal models for preclinical testing. Despite significant attempts to mitigate this problem, increasing evidence continues to suggest an imbalance between pharmaceutical investment and clinical impact. A major contributor to the problem lies in the disparity between human pathophysiology and their corresponding animal model. Coupled with rising costs, constrained time and limited resources, there is a need to develop more robust and reliable methods for chronic wound drug discovery and development.
In relation to chronic wounds, animal models are essential for studying the pathophysiology of these wounds. Importantly, these models serve as testing platforms for the identification of beneficial topical, systemic, and device-based therapeutics. However, the absence of a true chronic wound model in animals has hindered the development of therapeutic compounds. In a Food and Drug Administration (FDA) published guidance for chronic cutaneous ulcers, it has been recognized that there is still a lack of ideal chronic wound models[117]. Despite the success of the diabetic wound models in identifying the therapeutic potential of numerous growth factors, few treatments have received FDA approval for efficacy. Thus far, Becaplermin gel (Regranex®, Smith and Nephew) is the only chronic wound therapeutic to have received FDA approval following completion of human clinical trials[118]. Hence, while diabetic wound models are widely adopted for chronic wound therapeutic testing, their relevance and efficacy for human chronic wounds remains debatable.
Chronic wounds do not occur in animals in the same manner as human[59]. Thoroughbred horses are the only known animals to suffer from chronic wounds with similar features to human chronic wounds[6]. Equine limbs, specifically distal limb wounds, often suffer from delayed wound healing. These wounds can arise from traumatic incidents or surgical interventions and become chronic[119]. Similar to human chronic ulcers, these wounds do not progress sequentially through the normal healing process. They have a weak yet prolonged inflammatory response. In addition, horses can suffer from tissue ischemia[120]. Local hypoxia at wounds eventually leads to the development of exuberant granulation tissue[120]. Ultimately, re-epithelialization and wound contraction is impeded[119]. Given the similarity to human skin architecture, thoroughbred horses could be an alternative model for the study of non-healing wounds in large animals[6]. However, the high economic cost of utilizing horses, alongside with ethical concerns, limits the feasibility of adopting horses as a model for preclinical therapeutic trials.
Development of delayed wound models is also complicated by the multifactorial nature of human chronic wounds [1,2]. As such, there is a strong demand for the optimal selection of models that best represent human chronic wounds. Various factors should be considered when determining the most appropriate animal model for testing wound healing therapeutics. Some factors include reproducibility, physiological relevance to human chronic wounds, tractability, and cost effectiveness. As described in the previous section (Section 4), many studies have concentrated exclusively on isolating different aspects of chronic ulcers, such as diabetes and IR, in a reproducible and cost-effective approach. Accordingly, therapeutics targeting specific aspects of human chronic wounds would be inclined to selecting a wound model with the corresponding dysregulated feature (e.g. anti-bacterial product would be tested on a bacteria-infected wound model). However, the use of a single therapeutic agent has so far proven insufficient in its clinical efficacy. This is hardly surprising given the many factors that can contribute to the complexity of chronic wound pathophysiology. At present, many delayed wound healing models fail to fully recapitulate key histological characteristics of human chronic wounds, such as delayed re-epithelialization, persistent inflammation, and high levels of senescent cells (Table 1). Accordingly, this could limit the predictive value of these animal models in the translation of chronic wound therapeutics. Alternatively, other studies have employed a combination of different chronic wound elements[7]. However, advancements in these combinatorial approaches have been restricted by the need to ensure animal welfare. Ethically, to have an open wound on animals for months or years would not be acceptable.
An ideal delayed wound model should incorporate key characteristics of human chronic wounds. These include features such as delayed re-epithelialization, hyper thickened non-migratory wound edges with overexpression of the gap junction protein connexin 43, persistent inflammation, elevated ROS levels, alkaline wound environment, excessive ECM degradation at wound edges, disrupted/impaired vasculature and sustained presence of senescent cells (Figure 2). Concurrently, the gene expression profiles of wounds from this model should also be validated to reflect trends similar to human chronic wounds. In particular, expression of proinflammatory genes and genes regulating protease activity should be significantly upregulated compared to acute wounds. Conversely, the levels of genes regulating anti-inflammatory cytokines and protease inhibitors should be downregulated. Genes involved in regulating the cell cycle checkpoints would reflect the extent of senescence in the wound. In addition, the clinical relevance of the model could be further improved by including other chronic wound causal factors such as ageing, diabetes and ischemic conditions.
An alternative approach to induce chronic wound features on animals could involve the use of materials known to hinder reepithelialization, impede wound contraction, promote chronic inflammation and/or induce hypoxia. Double flanged silicon blocks inserted into wounds for 1–3 weeks cause tissue ischemia[121]. These wounds also suffer from delayed wound closure and persistent inflammation[121]. However, this method involves surgical procedures to secure the placement of silicon blocks at the wound site. Hence, complications could arise from infection at the incision site. The complexity of surgical procedures could also limit the accessibility and use of this model.
The ideal delayed wound model should be reproducible in both small and large animals i.e. both rodents and pigs. Of note, pigs have similar skin architecture to humans[59]. Unlike rodents, wounds on pigs heal largely by re-epithelialization and not contraction, which is similar to humans[59]. However, the cost involved to conduct wound healing studies on pigs is at least 20 times higher than similar studies on rodents. Hence, preliminary wound healing therapeutic pre-clinical studies, establishing treatment regime and therapeutic concentration, should be conducted on rodents first before moving onto more costly pig wound models. Prior to commencing clinical trials, there must be supporting data from at least 2 different species, reemphasizing the importance of replicating the wound models on both rodents and pigs[122].
Preclinical animal testing is a requirement for any novel therapeutic prior to entering human clinical trials. However, rising costs and high failure rates have led to the re-evaluation of the value of animal studies. Advancement in technologies have led to many companies incorporating human in silico trials to refine and reduce the use of animals[123]. Briefly, in silico clinical trials utilize computer simulations and modeling of human systems to predict various outcomes such as toxicity and efficacy[123]. In recent years, these in silico trials have been accepted as evidence by different regulatory agencies such as the FDA and European Medicines Agency upon assessment of the credibility of the predictive model used [124]. Nonetheless, a one-to-one replacement of animal testing with in silico trials is generally not practicable due to the complexity of the human physiology. Instead, a combination of both animal and in silico predictive models could contribute to a better mechanistic understanding of the safety and efficacy of a new drug with the human biological system.
Ultimately, to expedite the development of novel chronic wound therapeutics, there is an urgent need to develop more robust and reproducible models that mimic human chronic wound pathologies. Until then, the lack of therapies for chronic wounds will remain a significant worldwide healthcare burden.
Funding
This research is supported by the Agency for Science, Technology and Research (A*STAR) under its Industry Alignment Fund – Pre-Positioning Programme (IAF-PP) grant number H17/01/a0/0C9 as part of the Wound Care Innovation for the Tropics (WCIT) Programme. This research is also supported by the Agency for Science, Technology and Research (A*STAR) under its Industry Alignment Fund – Pre-Positioning Programme (IAF-PP) grant number H1701a0004 and the Skin Research Institute of Singapore, Phase 2: SRIS@Novena. Nanyang Technological University (Start-up grant) and the Ministry of Education (Tier 1 T1-002-098 and T1-002-013) also supported this research.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as: • of interest. •• of considerable interest
1. Guo S, DiPietro LA. Factors affecting wound healing. J Dent Res. 2010;89(3):219–229.
2. Frykberg RG, Banks J. Challenges in the Treatment of Chronic Wounds. Adv Wound Care. 2015;4(9):560–582.
3. Richmond NA, Maderal AD, Vivas AC. Evidence-based management of common chronic lower extremity ulcers. Dermatol Ther. 2013;26 (3):187–196.
4. Mustoe TA, O’Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Reconstr Surg. 2006;117:35–41.
5. Shrimanker M, Patel N, Modi H, et al. A review : screening models for wound healing activity in animals. Am J Pharmtech Res. 2013;3:238–251.
6. Harman RM, Theoret CL, Van De Walle GR. The horse as a model for the study of cutaneous wound healing. Adv Wound Care. 2021;10 (7):381–399.
A useful article that demonstrates how the horse is a physiologically relevant model that can be used in the study of wound healing. This article discusses the similarities in skin architecture between humans and horses and how this contributes to differences in wound healing processes. In addition, naturally occurring chronic skin pathologies in horses are highlighted.
7. Nunan R, Harding KG, Martin P. Clinical challenges of chronic wounds: searching for an optimal animal model to recapitulate their complexity. DMM Dis Model Mech. 2014;7(11):1205–1213.
8. Mustoe T. Understanding chronic wounds: a unifying hypothesis on their pathogenesis and implications for therapy. Am J Surg. 2004;187(5):S65–S70.
9. Sen CK. Human wound and its burden: updated 2020 compendium of estimates. Adv Wound Care. 2021;10(5):281–292.
A comprehensive review that provides an update of how chronic wounds are impacting the healthcare system. In addition to cellular and molecular events that impact wound healing, this article also elaborates on the social determinants contributing to chronic wounds
10. Sen CK. Human wounds and its burden: an updated compendium of estimates. Adv Wound Care. 2019;8(2):39–48.
11. Roth RS, Lowery JC, Hamill JB. Assessing persistent pain and its relation to affective distress, depressive symptoms, and pain catastrophizing in patients with chronic wounds: a pilot study. Am J Phys Med Rehabil. 2004;83(11):827–834.
12. Farag YMK, Gaballa MR. Diabesity: an overview of a rising epidemic. Nephrol Dial Transplant. 2011;26(1):28–35.
13. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6 (265):265sr6.
14. Singer AJ, Clark RA. Cutaneous wound healing. NEnglJMed. 1999;341(10):738–746.
15. Chen D, Hao H, Fu X, et al. Insight into reepithelialization: how do mesenchymal stem cells perform? Stem Cells Int. 2016;1–9.
16. Levy V, Lindon C, Zheng Y, et al. Epidermal stem cells arise from the hair follicle after wounding. FASEB J. 2007;21(7):1358–1366.
17. Goliger JA, Paul DL. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. MolBiol Cell. 1995;6(11):1491–1501.
18. Werner S, Smola H, Liao X, et al. The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science. 1994;266(80):819–822.
19. Rousselle P, Braye F, Dayan G. Re-epithelialization of adult skin wounds: cellular mechanisms and therapeutic strategies. Adv Drug Deliv Rev. 2019;146:344–365.
20. Usui ML, Mansbridge JN, Carter WG, et al. Keratinocyte migration, proliferation, and differentiation in chronic ulcers from patients with diabetes and normal wounds. J Histochem Cytochem. 2008;56(7):687–696.
21. Stojadinovic O, Pastar I, Vukelic S, et al. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med. 2008;12(6b):2675–2690.
22. Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127 (3):514–525.
23. Liu YC, Margolis DJ, Isseroff RR. Does inflammation have a role in the pathogenesis of venous ulcers: a critical review of the evidence. J Invest Dermatol. 2011;131(4):818–827.
24. Khanna S, Biswas S, Shang Y, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;4(3):e9539.
25. Rosen A, Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 1999;6(1):6–12.
26. Zhao R, Liang H, Clarke E, et al. Inflammation in chronic wounds. Int J Mol Sci. 2016;17(12):2085.
27. Harris IR, Yee KC, Walters CE, et al. Cytokine and protease levels in healing and non-healing chronic venous leg ulcers. Exp Dermatol. 1995;4(6):342–349.
28. Sutcliffe JES, Thrasivoulou C, Serena TE, et al. Changes in the extracellular matrix surrounding human chronic wounds revealed by 2-photon imaging. Int Wound J. 2017;14(6):1225–1236.
29. Caley MP, Martins VLC, O’Toole EA. Metalloproteinases and wound healing. Adv Wound Care. 2015;4(4):225–234.
30. Yager DR, Nwomeh BC. The proteolytic environment of chronic wounds. Wound Repair Regen. 1999;7(6):433–441.
31. Trengove NJ, Stacey MC, Macauley S, et al. Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen. 1999;7(6):442–452.
32. Yager DR, Zhang LY, Liang HX, et al. Wound fluids from human pressure ulcers contain elevated matrix metalloproteinase levels and activity compared to surgical wound fluids. J Invest Dermatol. 1996;107(5):743–748.
33. Bullen EC, Longaker MT, Updike DL, et al. Tissue inhibitor of metalloproteinase-1 is decreased and activated gelatinases are increased in chronic wounds. J Invest Dermatol. 1995;104(2):236–240.
34. Harding KG, Moore K, Phillips TJ. Wound chronicity and fibroblast senescence - Implications for treatment. Int Wound J. 2005;2 (4):364–368.
35. Neidernhofer L. DNA damage, cellular senescence in health and disease. Innov Aging. 2020;4(Supplement_1):742.
36. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–733.
37. Wilkinson HN, Hardman MJ. Senescence in wound repair: emerging strategies to target chronic healing wounds. Front Cell Dev Biol. 2020;8. DOI:10.3389/fcell.2020.00773.
38. Lopes-Paciencia S, Saint-Germain E, Rowell MC, et al. The senescence-associated secretory phenotype and its regulation. Cytokine. 2019;117:15–22.
39. Castilla DM, Liu Z-J, Velazquez OC. Oxygen: implications for wound healing. Adv Wound Care. 2012;1(6):225–230.
40. Soneja A, Drews M, Malinski T. Role of nitric oxide, nitroxidative and oxidative stress in wound Pharmacol 2005;57:108–119.
41. Rodriguez PG, Felix FN, Woodley DT, et al. The role of oxygen in wound healing: a review of the literature. Dermatologic Surg. 2008;34:1159–1169.
42. Schreml S, Szeimies RM, Prantl L, et al. Oxygen in acute and chronic wound healing. Br J Dermatol. 2010;163(2):257–268.
43. Jünger M, Steins A, Hahn M, et al. Microcirculatory dysfunction in chronic venous insufficiency (CVI). Microcirculation. 2000;7(6):3–12.
44. Eming SA. Increased levels of the soluble variant of the vascular endothelial growth factor receptor VEGFR-1 are associated with a poor prognosis in wound healing. J Invest Dermatol. 2004;123 (4):799–802.
45. Segal AW. How superoxide production by neutrophil leukocytes kills microbes. Innate Immun to Pulm Infect. 2008;279:92–98.
46. Schäfer M, Werner S. Oxidative stress in normal and impaired wound repair. Pharmacol Res. 2008;58(2):165–171.
47. Siddiqui AR, Bernstein JM. Chronic wound infection: facts and controversies. Clin Dermatol. 2010;28(5):519–526.
48. James GA, Swogger E, Wolcott R, et al. Biofilms in chronic wounds. Wound Repair Regen. 2008;16(1):37–44.
49. Høiby N, Bjarnsholt T, Givskov M, et al. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents. 2010;35(4):322–332.
50. Fazli M, Bjarnsholt T, Kirketerp-Møller K, et al. Quantitative analysis of the cellular inflammatory response against biofilm bacteria in chronic wounds. Wound Repair Regen. 2011;19(3):387–391.
51. Sanchez MC, Lancel S, Boulanger E, et al. Targeting oxidative stress and mitochondrial dysfunction in the treatment of impaired wound healing: a systematic review. Antioxidants. 2018;7(8):98.
52. Yang P, Pei Q, Yu T, et al. Compromised wound healing in ischemic type 2 diabetic rats. PLoS One. 2016;11(3):e0152068.
53. Chen C, Schultz GS, Bloch M, et al. Molecular and mechanistic validation of delayed healing rat wounds as a model for human chronic wounds. Wound Repair Regen. 1999;7(6):486–494.
54. Roy S, Biswas S, Khanna S, et al. Characterization of a preclinical model of chronic ischemic wound. Physiol Genomics. 2009;37 (3):211–224.
55. Liechty KW, Crombleholme TM, Quinn TM, et al. Elevated platelet-derived growth factor-B in congenital cystic adenomatoid malformations requiring fetal resection. J Pediatr Surg. 1999;34 (5):805–809.
56. Corral CJ, Siddiqui A, Wu L, et al. Vascular endothelial growth factor is more important than basic fibroblastic growth factor during ischemic wound healing. Arch Surg. 1999;134(2):200–205.
57. Ahn ST, Mustoe TA. Effects of ischemia on ulcer wound healing: a new model in the rabbit ear. Ann Plast Surg. 1990;24(1):17–23.
58. Steinberg JP, Hong SJ, Geringer MR, et al. Equivalent effects of topically-delivered adipose-derived stem cells and dermal fibroblasts in the ischemic rabbit ear model for chronic wounds. Aesthetic Surg J. 2012;32(4):504–519.
59. Grada A, Mervis J, Falanga V. Research techniques made simple: animal models of wound healing. J Invest Dermatol. 2018;138 (10):2095–2105.
A concise review that discusses animal models that are used in both acute and impaired wound healing experiments. This article also describes the differences between the skin architecture of different animals and how this compares to human skin
60. Trujillo AN, Kesl SL, Sherwood J, et al. Demonstration of the rat ischemic skin wound model. J Vis Exp. 2015;98:e52637.
61. Hsueh YY, Wang DH, Huang TC, et al. Novel skin chamber for rat ischemic flap studies in regenerative wound repair. Stem Cell Res Ther. 2016;7(1). DOI:10.1186/s13287-016-0333-0
62. Schäffer M, Witte M, Becker HD. Models to study ischemia in chronic wounds. Int J Low Extrem Wounds. 2002;1(2):104–111.
63. Gould LJ, Leong M, Sonstein J, et al. Optimization and validation of an ischemic wound model. Wound Repair Regen. 2005;13 (6):576–582.
64. Myers WT, Gould LJ. Animal models of tissue ischemia to evaluate the importance of oxygen in the wound healing environment. Wounds. 2008;20(1):9–17.
65. Woo K, Marin J, Brandys T. Assessing chronic wound perfusion in the lower extremity: current and emerging approaches. Chronic Wound Care Manag Res. 2015;2:149–157.
66. Patil P, Martin JR, Sarett SM, et al. Porcine ischemic wound-healing model for preclinical testing of degradable biomaterials. Tissue Eng - Part C Methods. 2017;23(11):754–762.
67. Edsberg LE, Black JM, Goldberg M, et al. Revised national pressure ulcer advisory panel pressure injury staging system. J Wound, Ostomy Cont Nurs. 2016;43(6):585–597.
68. Jaul E, Barron J, Rosenzweig JP, et al. An overview of co-morbidities and the development of pressure ulcers among older adults. BMC Geriatr. 2018;18(1):305.
69. Smith DM. Pressure ulcers in the nursing home. Ann Intern Med.1995;123(6):433–442.
70. Şener G, Sert G, Özer Şehirli A, et al. Melatonin protects against pressure ulcer-induced oxidative injury of the skin and remote organs in rats. J Pineal Res. 2006;40(3):280–287.
71. Tong M, Tuk B, Hekking IM, et al. Heparan sulfate glycosaminoglycan mimetic improves pressure ulcer healing in a rat model of cutaneous ischemia-reperfusion injury. Wound Repair Regen. 2011;19(4):505–514.
72. Peirce SM, Skalak TC, Rodeheaver GT. Ischemia-reperfusion injury in chronic pressure ulcer formation: a skin model in the rat. Wound Repair Regen. 2000;8(1):68–76.
73. Kwek MSY, Thangaveloo M, Hui SLB, et al. Characterisation of an ischemia reperfusion model for the formation of a stage I pressure ulcer in mouse skin. J Tissue Viability. 2021;30(3):352–362.
74. Tatlidede S, McCormack MC, Eberlin KR, et al. A novel murine island skin flap for ischemic preconditioning. J Surg Res. 2009;1(1):112–117.
75. Edsberg LE. Pressure ulcer tissue histology: an appraisal of current knowledge. Ostomy Wound Manag. 2007;53:40–49.
76. Strong AL, Bowles AC, MacCrimmon CP, et al. Characterization of a murine pressure ulcer model to assess efficacy of adipose-derived stromal cells. Plast Reconstr Surg - Glob Open. 2015;3(3):e334.
77. Nguyen PKT, Smith AL, Reynolds KJ. A literature review of different pressure ulcer models from 1942-2005 and the development of an ideal animal model. Australas Phys Eng Sci Med. 2008;31(3):223–225.
78. King A, Austin A. Animal models of type 1 and type 2 diabetes mellitus. Anim Model Study Hum Dis Second Ed. 2017:245–265
79. King AJF. The use of animal models in diabetes research. Br J Pharmacol. 2012;166(3):877–894.
A comprehensive review that outlines models currently used in diabetic research. This includes those used to model type 1 or type 2 diabetes with transgenic and knock-out mice models or through chemical induction. Furthermore, this article discusses the factors that one can consider when choosing an appropriate animal model for diabetes research.
80. Davis FM, Kimball A, Boniakowski A, et al. Dysfunctional Wound Healing in Diabetic Foot Ulcers: new Crossroads. Curr Diab Rep. 2018;18(4):18.
81. Jeffcoate WJ, Vileikyte L, Boyko EJ, et al. Current challenges and opportunities in the prevention and management of diabetic foot ulcers. Diabetes Care. 2018;41(4):645–652.
82. Ang Y, Yap CW, Saxena N, et al. Diabetes-related lower extremity amputations in Singapore. Proc Singapore Healthc. 2017;26 (2):76–80.
83. Wolf E, Braun-Reichhart C, Streckel E, et al., Genetically engineered pig models for diabetes research. Transgenic Res. 2014;23(1):27–38.
A useful review that describes existing genetically engineered pig models and how they mimic the human disease mechanisms of diabetes at the molecular level. In addition, this article also discusses the potential applications for these models.
84. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–25.
85. Chan RK, Liu PH, Pietramaggiori G, et al. Effect of recombinant platelet-derived growth factor (Regranex®) on wound closure in genetically diabetic mice. J Burn Care Res. 2006;27(2):202–205.
86. Saaristo A, Tammela T, Farkkila A, et al. Vascular endothelial growth factor-C accelerates diabetic wound healing. Am J Pathol. 2006;169 (3):1080–1087.
87. Mohammed-Ali Z, Carlisle RE, Nademi S, et al. Animal models of kidney disease. Anim Model Study Hum Dis Second Ed 2017;379–417
88. Michaels J, Churgin SS, Blechman KM, et al. db/db mice exhibit severe wound-healing impairments compared with other murine diabetic strains in a silicone-splinted excisional wound model. Wound Repair Regen. 2007;15(5):665–670.
89. Ritskes-Hoitinga M, Tobin G, Jensen TL, et al. Nutrition of the laboratory mouse. Lab Mouse. 2012. DOI:10.1016/B978-0-12- 382008-2.00024-6
90. Galiano RD, J MV, Dobryansky M, et al. Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen. 2004;12(4):485–492.
91. Wilkinson HN, Clowes C, Banyard KL, et al. Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. J Invest Dermatol. 2019;139(5):1171–1181.
92. Nguyen VT, Farman N, Palacios-Ramirez R, et al. Cutaneous wound healing in diabetic mice is improved by topical mineralocorticoid receptor blockade. J Invest Dermatol. 2020;140(1):223–234.
93. Leiter EH, Strobel M, O’Neill A, et al. Comparison of two new mouse models of polygenic type 2 diabetes at the Jackson laboratory, NONcNZO10Lt/J and TALLYHO/JngJ. J Diabetes Res. 2013;2013:1–7.
94. Daly JM. Limitations of the db/db mouse in translational wound healing research: is the NONcNZO10 polygenic mouse model superior? Yearb Surg. 2011;18:605–613.
An interesting study that investigates the difference in wound healing between different diabetic wound healing models. This article discusses the limitations of the db/db mouse model, which is one of the commonly used diabetic wound healing model. In addition, they highlight the clinical relevance of using polygenic animals in diabetic research.
95. Blaber SI, Diaz J, Blaber M. Accelerated healing in NONcNZO10/LtJ type 2 diabetic mice by FGF-1. Wound Repair Regen. 2015;23 (4):538–549.
96. Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. 2008;51(2):216–226.
97. Bacevic M, Rompen E, Radermecker R, et al. Practical considerations for reducing mortality rates in alloxan-induced diabetic rabbits. Heliyon. 2020;6(6):e04103.
98. Furman BL. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol. 2015;70(1):5.47.1–5.47.20.
99. Wang CM, Lincoln J, Cook JE, et al. Abnormal connexin expression underlies delayed wound healing in diabetic skin. Diabetes. 2007;56(11):2809–2817.
100. Ansell DM, Marsh C, Walker L, et al. Evaluating STZ-induced impaired wound healing in rats. J Invest Dermatol. 2018;138(4):994–997.
101. Velander P, Theopold C, Hirsch T, et al. Impaired wound healing in an acute diabetic pig model and the effects of local hyperglycemia. Wound Repair Regen. 2008;16(2):288–293.
102. Sharma AN, Elased KM, Garrett TL, et al. Neurobehavioral deficits in db/db diabetic mice. Physiol Behav. 2010;101(3):381–388.
103. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009;37:81–90.
104. Deeds MC, Anderson JM, Armstrong AS, et al. Single dose streptozotocin-induced diabetes: considerations for study design in islet transplantation models. Lab Anim. 2011;45(3):131–140.
105. Dufrane D, Van Steenberghe M, Guiot Y, et al. Streptozotocininduced diabetes in large animals (pigs/primates): role of GLUT2 transporter and β-cell plasticity. Transplantation. 2006;81(1):36–45.
106. Ganesh K, Sinha M, Mathew-Steiner SS, et al. Chronic wound biofilm model. Adv Wound Care. 2015;4(7):382–388.
107. Fila G, Kasimova K, Arenas Y, et al. Murine model imitating chronic wound infections for evaluation of antimicrobial photodynamic therapy efficacy. Front Microbiol. 2016;9:1258.
108. Trøstrup H, Thomsen K, Christophersen LJ, et al. Pseudomonas aeruginosa biofilm aggravates skin inflammatory response in BALB/c mice in a novel chronic wound model. Wound Repair Regen. 2013;21(2):292–299.
109. Schierle CF, De La Garza M, Mustoe TA, et al. Staphylococcal biofilms impair wound healing by delaying reepithelialization in a murine cutaneous wound model. Wound Repair Regen. 2009;17(3):354–359.
110. Brandenburg KS, Calderon DF, Kierski PR, et al. Novel murine model for delayed wound healing using a biological wound dressing with Pseudomonas aeruginosa biofilms. Microb Pathog. 2018;122:30–38.
111. Gurjala AN, Geringer MR, Seth AK, et al. Development of a novel, highly quantitative in vivo model for the study of biofilm-impaired cutaneous wound healing. Wound Repair Regen. 2011;19(3):400–410.
112. Shukla SK, Sharma AK, Gupta V, et al. Challenges with wound infection models in drug development. Curr Drug Targets. 2020;21(13):1301–1312.
113. Burmølle M, Thomsen TR, Fazli M, et al. Biofilms in chronic infections - A matter of opportunity - Monospecies biofilms in multispecies infections. FEMS Immunol Med Microbiol. 2010;59 (3):324–336.
114. Pletzer D, Mansour SC, Wuerth K, et al. New mouse model for chronic infections by gram-negative bacteria enabling the study of anti-infective efficacy and host-microbe interactions. MBio. 2017 Feb 28;8:1.
115. Zhao G, Hochwalt PC, Usui ML, et al. Delayed wound healing in diabetic (db/db) mice with Pseudomonas aeruginosa biofilm challenge: a model for the study of chronic wounds. Wound Repair Regen. 2010;18(5):467–477.
116. Van Norman GA. Limitations of animal studies for predicting toxicity in clinical trials: is it time to rethink our current approach? JACC Basic to Transl Sci. 2019;4(7):845–854.
A useful review that discusses issues in the value of using animal models in preclinical studies to predict potential toxicity and efficacy of new therapeutics. This review also suggests alternatives to the use of animals.
117. Guidance for industry: chronic cutaneous ulcer and burn wounds -Developing products for treatment: draft - Not for implementation. Wound Repair Regen. 2001;9(4):258–268.
An FDA published guide stating that there is still a lack of reliable chronic wound models
118. Ansell DM, Holden KA, Hardman MJ. Animal models of wound repair: are they cutting it? Exp Dermatol. 2012;21(8):581–585.
119. Westgate SJ, Percival SL, Knottenbelt DC, et al. Chronic equine wounds: what is the role of infection and biofilms? Wounds. 2010;22(6):138–145.
120. Jørgensen E, Bay L, Skovgaard LT, et al. An equine wound model to study effects of bacterial aggregates on wound healing. Adv Wound Care. 2019;8(10):487–498.
121. Jung Y, Son D, Kwon S, et al., Experimental pig model of clinically relevant wound healing delay by intrinsic factors. Int Wound J. 2013;10(3):295–305.
An interesting porcine study that involves the use of materials to induce certain chronic wound features. They demonstrate that ischemia can be induced through the use of double flanged silicon blocks. In addition, healing is delayed, and wounds suffer from persistent inflammation.
122. Prior H, Baldrick P, Beken S, et al. Opportunities for use of one species for longer-term toxicology testing during drug development: a cross-industry evaluation. Regul Toxicol Pharmacol. 2020;113:104624.
123. Madden JC, Enoch SJ, Paini A, et al. A review of in silico tools as alternatives to animal testing: principles, resources and applications. Altern Lab Anim. 2020;48(4):146–172.
124. Viceconti M, Pappalardo F, Rodriguez B, et al. In silico trials: verification, validation and uncertainty quantification of predictive models used in the regulatory evaluation of biomedical products. Methods. 2021;185:120–127.
CONTACT David L. Becker
该Email地址已收到反垃圾邮件插件保护。要显示它您需要在浏览器中启用JavaScript。 Lee Kong Chian School of Medicine, Nanyang Technological University 11 Mandalay Road #17-01,308232, Singapore# These authors contributed equally to this work.
EXPERT OPINION ON DRUG DISCOVERY
https://doi.org/10.1080/17460441.2023.2158809
© 2022 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives License (http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the original work is properly cited, and is not altered, transformed, or built upon in any way.
This article is excerpted from the EXPERT OPINION ON DRUG DISCOVERY by Wound World.

