
伤口世界

- 星期一, 08 6月 2026
Comprehensive Approaches to Diagnosis and Treatment of Sensitive Skin
Hye One Kim 1,* , Ji Young Um 1,* , Han Bi Kim 1 , So Yeon Lee 1 , Hyun Choi 2 , Jihye Kim 3 , Eunbi Ko 3 , Bo Young Chung 1 , Chun Wook Park 1
1 Department of Dermatology, Hallym University Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea
2 Make-up Lab, R&D Center, Kolmar Korea, Seoul, Korea
3 Clinical Research Lab, Amorepacific R&I Center, Yongin, Korea
ABSTRACT
Sensitive skin (SS) is increasingly recognized as a complex syndrome characterized by discom-fort and heightened sensitivity to otherwise harmless stimuli, such as environmental changes, physical contact, and cosmetic products. This condition poses challenges in both diagnosis and treatment due to its variable presentation and subjective nature. The pathophysiological features of SS include neurogenic inflammation and small fiber neuropathy, largely driven by the hyperactivation of sensory nerves. This hyperactivation is closely associated with transient receptor potential (TRP) channels, particularly TRPV1, which contribute to the exaggerated sensory responses seen in SS. Furthermore, psychological factors like stress and anxiety, along with environmental stressors such as pollution and ultraviolet exposure, play significant roles in exacerbating symptoms. The diverse and individualized responses to stimuli make it difficult to establish standardized diagnostic criteria for SS, necessitating a combination of subjective diagnostic tools (e.g., the Sensitive Scale-10) and objective assessments (e.g., transepidermal water loss and lactic acid sting test) to accurately identify and assess SS. This paper provides a comprehensive review of SS, covering its definition, prevalence, pathogenesis, diagnostic challenges, and management strategies, and highlights the importance of personalized care in effectively managing SS and improving patient quality of life.
Keywords: Skin diseases; Skin irritancy tests; Skin physiological phenomena; Pruritus; Pathophysiology; Sensitive Scale-10; Sensitive skin

- 星期六, 06 6月 2026
Cai’s herbal tea enhances mitochondrial autophagy of type 1 diabetic mellitus β cells through the AMPK/mTOR pathway and alleviates inflammatory response
Hongchun Li1 ·Yanfei Gao2 · Mengdi Li1 ·Yue Dong1 · Jie Chen3 · Bingyue Zhang3 · Kaiqiang Li4 ·Yuqun Cai3
Received: 11 January 2024 / Accepted: 30 May 2024 / Published online: 2 July 2024©The Author(s) 2024
Abstract
Background This study investigates the therapeutic mechanisms of Cai’s Herbal Tea in Type 1 Diabetes Mellitus (T1DM) mice,focusing on its effects on mitochondrial change and autophagy via the AMP-activated protein kinase (AMPK)—mammalian target of rapamycin (mTOR) pathway.
Methods The composition of Cai’s Herbal Tea was analyzed by Ultra-High Performance Liquid Chromatography-Quadrupole Time of Flight Mass Spectrometry (UHPLC-Q/TOF-MS). C57BL/6 mice and Min6 pancreatic beta cells were divided into control, diabetic mellitus (DM)/high glucose (HG), and treatment groups (low, medium, and high doses of Cai’s Tea, and Metformin). Key physiological parameters, pancreatic islet health, Min6 cell morphology, viability, and insulin (INS) secretion were assessed. Small Interfering RNA-AMPK (si-AMPK) was utilized to confirm the pathway involvement. Results Cai’s Herbal Tea improved body weight, pancreatic islet pathological injury, and INS secretion whereas reduced total triglycerides, fasting blood sugar, and Interferon gamma (INF-γ) in T1DM mice,particularly at higher doses. In Min6 cells, Cai’s Tea mitigated HG-induced damage and proinflammatory response, enhancing cell viability and INS secretion. Notably, it reduced swelling and improved cristae structure in treated groups of mitochondria and promoted autophagy via the AMPK-mTOR pathway, evidenced by increased LC3II/LC3I and P-AMPK/AMPK ratios, and decreased P-mTOR/ mTOR and P62 expressions in pancreatic islet β-cells.Furthermore,these effects were converted by si-AMPK interference. Conclusion Cai’s Herbal Tea exhibits significant therapeutic efficacy in T1DM mice by improving mitochondrial health and inducing autophagy through the AMPK-mTOR pathway in pancreatic islet β-cells. These findings highlight its potential as a therapeutic approach for T1DM management.
Keywords Animal study · Cai’s herbal tea · Type 1 diabetes mellitus · AMPK-mTOR pathway · Autophagy
Abbreviations
T1DM Type 1 diabetes mellitus
INS Insulin
IL-1β Interleukin-1β
MAP1LC3
AMPK
UPLC-Q/TOF-MS
Microtubule-associated protein 1 light chain 3
AMP-activated protein kinase Ultra performance liquid chromatography-quadrupole/time of flight-mass
spectrometry
IDA Information dependent acquisition
TG Total triglycerides
TC Total cholesterol
ELISA Enzyme-linked immunosorbent assay
INF-γ Interferon gamma
IL-4 Interleukin 4
HE Hematoxylin and eosin
IHC Immunohistochemistry
TEM Transmission electron microscopy
DMEM Dulbecco’s modified eagle medium
HG High-glucose
si-NC SiRNA negative control
si-AMPK SiRNA AMP-activated protein
kinase
MTT 3-(4,5-Dimethylthiazol-2-yl)-2,5-di-
phenyltetrazolium bromide
GSIS Glucose stimulated INS secretion
BIS Basal INS secretion
ISI INS secretion index
NOD Non-obese diabetic

- 星期五, 05 6月 2026
Silencing the FABP3 gene in insulin‑secreting cells reduces fatty acid uptake and protects against lipotoxicity
Ayman Hyder1 · Basma Sheta1 · Manar Eissa1 · Jürgen Schrezenmeir2
Received: 20 February 2024 / Accepted: 18 June 2024 / Published online: 4 July 2024©The Author(s) 2024
Abstract
Background Long-term exposure of pancreatic islets to fatty acids (FAs), common in obesity, metabolic syndrome, and type 2 diabetes, leads to a compensatory hyperactivity followed by inflammation, apoptosis, dysfunctional beta cells, and results in insulin dependence of the patient.Restriction of fatty uptake by islet beta cells may protect them from lipotoxicity. Purpose Pancreatic islet beta cells express the fatty acid binding protein 3 (FABP3) to bind FAs and to orchestrate lipid signals.Based on this,we investigated whether downregulation ofFABP3,by Fabp3 silencing,might slow lipid metabolism and protect against lipotoxicity in insulin-secreting cells.
Results Neither Fabp3 silencing, nor overexpression affected the glucose-stimulated insulin secretion in absence of FAs. Fabp3 silencing decreased FA-uptake,lipid droplets formation, and the expression of the lipid accumulation-regulating gene Dgat1 in Ins1E cells.It reduced FA-induced inflammation by deactivation of NF-κB,which was associated with upregulation of IκBα and deactivation of the NF-κB p65 nuclear translocation, and the downregulation of the cytokines ILl-6,IL-1β, and TNFα.Ins1E cells were protected from the FA-induced apoptosis as assessed by different parameters including DNA degradation and cleaved caspase-3 immunoblotting.Furthermore,FABP3 silencing improved the viability,Pdx1 gene expression, and the insulin-secreting function in cells long-term cultured with palmitic acid.All results were confirmed by the opposite action rendered by FABP3 overexpression.
Conclusion The present data reveals that pancreatic beta cells can be protected from lipotoxicity by inhibition of FA-uptake, intracellular utilization and accumulation. FABP3 inhibition, hence, may be a useful pharmaceutical approach in obesity, metabolic syndrome, and type 2 diabetes.
Keywords Insulin secretion · Beta cell · Fatty acid · Binding protein · Inflammation · Apoptosis
Abbreviations
FA Fatty acid
FABP Fatty acid binding protein
GSIS Glucose-stimulated insulin secretion HFD High-fat diet
Il Interleukin
NF-κB Nuclear factor kappa BPUFA Polyunsaturated fatty acid
TG Triglycerides
TNFa Tumor necrosis factor alpha
Introduction
Pancreatic islet beta cell lipotoxicity refers to the excessive accumulation of FAs within cells leading to their dysfunction, death by apoptosis [1]. There is a strong correlation between lipotoxicity-induced beta cell dysfunction and death and the development of obesity, metabolic syndrome, and progression of type 2 diabetes [2].These disorders are linked with increased oxidative stress, ceramide accumulation, and activation of stress responses [3]. In pancreatic islets of Langerhans, FAs have a dual effect [4], since the shortterm exposure stimulates insulin secretion, while the longterm exposure to FAs increases basal insulin release and reduces glucose-stimulated insulin secretion (GSIS). It also
causes islet beta cell failure and apoptosis, especially when glucose levels are elevated. The contribution of lipotoxicity to deterioration of insulin secretion and peripheral insulin resistance, and its association with the increased obesity prevalence, metabolic syndrome, and type 2 diabetes were early reported [5]. There is evidence that the 5- to 10-fold increment in islet lipid content that occurs in the prediabetic phase triggers the compensatory beta cell hyperplasia and hyperinsulinemia. Further elevation in islets’ fats reverses the preceding compensatory changes and initiates beta cell dysfunction and diabetes [6].
The transport and metabolism of the insoluble FAs are regulated by membrane-associated and cytosolic proteins that bind and transport FAs.There are ten distinct fatty acid binding proteins (FABPs), with tissue-specific expression patterns [7]. These intracellular FA transport proteins participate in lipid metabolism by binding FAs,regulating gene expression, and orchestrating lipid signals.Pancreatic islets mostly express the heart/muscle type (FABP3)to bind,traffic and metabolize FAs [8]. FABP3 is a 14–15 kDa protein abundantly expressed in the muscle, heart, and brain [9]. It binds to long-chain FAs and transports them to different subcellular compartments for lipid storage and metabolism[10].In addition to its lipid metabolic role,FABP3 has other important functions on signaling transduction and transcriptional regulation [11, 12]. For example, overexpression ofFABP3 upregulated the phosphorylation of MAPK signaling pathway and decreased phosphorylated Akt levels, which may account for the augmentation of apoptosis after myocardial infarction [13]. Also, FABP3 is involved in the control of DNA methylation of the Gad67 promoter and activation of GABAergic neurons in the anterior cingulate cortex [14].
In different cell systems, variable FABPs were reported to boost FA uptake and triglycerides (TG) accumulation. Manipulation of different FABP family members has been reported to control FA uptake and intracellular lipid accumulation. In mouse liver, knocking down the fabp1 gene reduced hepatic TG accumulation and FA uptake [15]. In mouse brain, Fabp3 gene knockout has been reported to decrease arachidonic PUFA uptake and alter the phospholipid composition [16]. In L cells (a fibroblast cell line), overexpression of FABP1 and 2 increased phospholipid content and altered the fatty acid composition of phospholipids [17]. Similarly, knocking down fabp4 in mouse prevented the progression of insulin resistance and atherosclerosis [18], whereas its knocking out improved osteoarthritis induced by HFD in mice [19].
Therefore, inhibition of FABP3 in the pancreatic islets may protect against TG accumulation and the consequent impaired insulin secretory function and beta cell death.The main hypothesis in this work is that the knocking down Fabp3 in insulin-secreting cells will reduce FA uptake and the resultant lipotoxicity and prevent apoptosis of
insulin-producing cells. The limited expression pattern of FABPs [20] suggests a quite specific action on islet beta cells. Consequently, the aim of the present work was to investigate the effect of FABP3 manipulation by silencing or overexpression on fatty acid uptake into beta cells and their inflammatory response upon FA exposure.
Materials and methods Tissue culture
The rat insulin-secreting beta cell line Ins1E cells [21] were cultured as published before [8]. Pancreatic islets were isolated as described before [22–24]from control or 16 w-highfat fed male rats.The fat content in the control diet was 4.5%, while that in the high-fat diet was 40% [25].Ins1E cells (passages 30–50) and islets were cultured at 37 °C with 5% CO2 in RPMI-1640 Glutamax medium supplemented with 10% fetal calf serum, 10 mmol/L Hepes, 50 μmol/L 2-mercaptoethanol, 1 mmol/L sodium pyruvate, 100 IU penicillin/ml and 100 μg streptomycin/ml. For the different experiments, cells were cultured in 6-well plates until reaching 80–90% confluence.
For the preparation of fatty acids [26], a stock solution was prepared by dissolving FAs in ethanol to a final concentration of 1 mol/L. Aliquots of this stock solution were dissolved in 10%fatty acid-free bovine serum albumin (Merck) solution in additive-free RPMI medium) to a concentration of 10 mmol/L by incubation in ultrasonic bath. The BSAbound solution was then diluted in culture media.Cells were fasted for 2 h in supplementation-free RPMI1640 medium before different incubations.
Cloning of FABP3
The method cloning using the plasmid pCAGGS was reported before [27, 28].To extract and clone the full-length coding sequence ofFABP3,RNA was isolated from rat muscle. The following oligonucleotide primers were designed after examination the sequences for the restriction enzymes(Xho1 and EcoR1)that neither cut within the FABP3 nor the pCAGGS vector sequences. The forward primer sequence was 5 ′-CTGGAATTCATGGCGGACGCCTTTGTC-3 ′ which has a restriction site for EcoR1,and the reverse primer was 5′-CAGCTCGAGTCACGCCTCCTTCGTAAG-3′with a restriction site for Xho1 . The PCR products were ligated into the vector (T4-Ligase Rapid-Kit (Fermentas, Thermo Fisher Scientific), and clones with the proper full-length FABP3 inserts were selected for sequencing. The cloned FABP3 was identically aligned to the genomic sequences.
Transfection for FABP3 silencing/overexpression
Ins1E cells were seeded in 24-well plates at a density of4 ×104 cells per well in 1 ml of culture medium and transfections performed at 85%cell confluence.Prior to transfection, TurboFect (Fermentas, Thermo Fisher Scientific) and MEM (Invitrogen) were mixed and incubated at room temperature for 20 min. 100 ng DNA of the FABP3 construct was added to the prepared transfection medium in a ratio of 3 µl transfection medium to 1 ng DNA as recommended by the manufacturer. Control samples were treated similarly and transfected with the empty vector (pCAGGS). After 48 and 72 h post transfection, RNA was isolated, and reverse transcribed to cDNA.FABP3 gene overexpression was confirmed using quantitative RT-PCR and protein Western blot.
For the small interfering (siRNA) experiments, the OnTarget plus smart siRNA pool (Dharmacon L-100472 -01-0010, ThermoFisher Scientific) that targets 4 different sequences was applied to silence FABP3 . A non-targeting scrambled sequence (siScr) was applied as a negative control. Ins1E cells were reverse transfected with the siRNA pool using siPORT NeoFX Transfection Reagent (Invitrogen). siRNA was prepared in Opti-MEM serum-free medium(Gibco)by mixing 2 ul of the transfection reagent with 5 nM siRNA at RT for 10 min. Ins1E cells (4 ×104 cells per well) were cultured in 24-well plates containing siRNA transfection reagent complexes, to allow for transfection to occur during initial cell adherence.The medium was changed after24 h by the ordinary culture medium and incubated for additional 24 and 48 h to allow cell recovery.RNA was extracted and reverse transcribed to cDNA. FABP3 gene knockdown was confirmed by quantitative RT-PCR and protein Western blot. Confirmation and efficiency of transfections are shown in the supplementary figure S1 .
Quantitative real‑time PCR
Total RNA was extracted and 2 μg RNA was reverse transcribed to first strand complementary DNA as reported before [29]. Quantitative RT-PCR analysis was performed on a quantity of cDNA that corresponded to 20 ng RNA. Quantitative PCR analysis was performed using SYBR Green. The thermal cycling program was 10 min at 95 °C for enzyme activation, denaturation for 15 s at 95 °C, 60 s annealing at 60 °C. A dissociation curve was run for each product to verify the absence of primer dimers or nonspecific products.Different primer pairs used in the present study for studied genes are listed in the supplementary Table S1 . To normalize expression data, β-actin was used as a housekeeping and internal control gene. Relative quantification was performed by ΔΔCt method. Data were presented as fold change from the control,whose values were considered as 1.
Western blotting
Protein was extracted from cells with the aid of tissue lysis buffer (50 mmol/L Hepes, pH 7.5, 150 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 1.5 mmol/L MgCl2, 1 mmol/LEGTA, protease cocktail (Roche, one tablet/10 ml final buffer volume). Protein SDS-PAGE electrophoresis was performed using Pierce protein gels (Thermo Scientific) and blotted onto a PVDF membrane (Santa Cruz Biotechnology).The membrane was probed with the following antibodies: beta actin (Santa Cruz), cleaved caspase 3 (Cell Signaling), FABP3 (Hycult), histone H3 (Abcam), NF-κB p65 (Dianova).Detection of proteins was carried out by incubation with appropriate HRP-conjugated secondary antibodies and detected by ECL detection reagents (Thermo-Fisher Scientific).
Fatty acid uptake and lipid droplet assay
The QBT Fatty Acid Uptake and lipid droplet fluorescence Assay Kit (Molecular Devices, product # R8132), which employs a BODIPY-dodecanoic (lauric) acid fluorescent fatty acid analog.For kinetic FA uptake reading,Ins1E cells of different treatments were seeded in 96-well plates with104 cells per well in 100 µl of complete RPMI medium and incubated at 37 °C for 24 h. After washing, cells were incubated in serum-free medium for 1 h., which was replaced at the end of the incubation time with 100 µl of the kit fatty acid loading buffer.Real-time uptake kinetic readings in live cells were started immediately with a fluorescence plate bottom reader.For the end point reading mode,cells were incubated for 1h before reading.For lipid droplet imaging,plates were incubated overnight (37 °C, 5% CO2)before examination with an inverted fluorescence microscope.
MTT assay
Ins1E cells were seeded in 96-well plates with 5 ×104 cells per well. Cells of different treatments were incubated with0.1–0.5 mmol/L palmitic acid for 24–72 h. Cell viability was determined by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT, Sigma) according to the manufacturer instructions. Cell culture supernatants were aspirated from plates and MTT solution (1 mg/ml) was added. Plates were incubated with MTT for 30 min at37 °C. Dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals, and the absorbance was measured at570 nm, with a correction at 690 nm.
DNA fragmentation assay
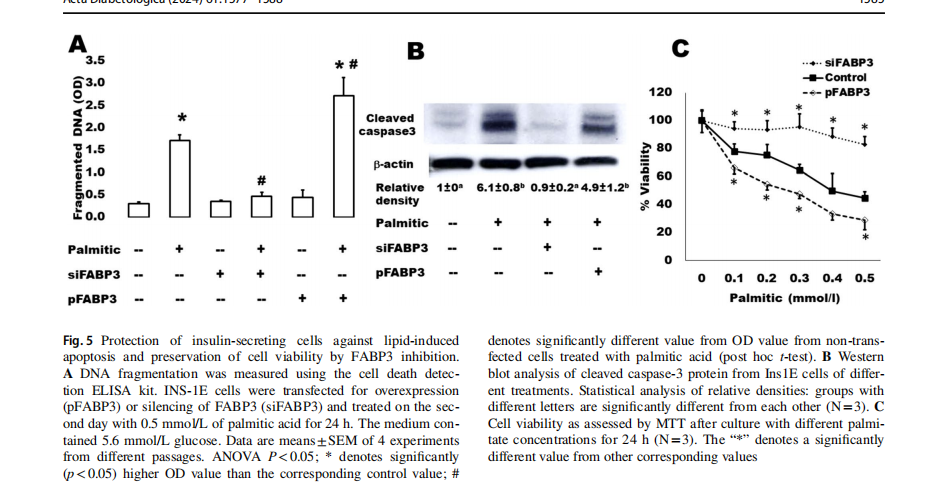
Cytoplasmic histone-associated DNA fragments were measured by the Cell Death Detection ELISA kit (Roche,Merck) following the manufacturer instructions. INS-1E cells were transfected for FABP3 overexpression or silencing and treated on the second day with 0.5 mmol/L of palmitic acid in the presence of 5.6 mmol/L glucose for 24 h. Cells were lysed as described by the kit manufacturer. After centrifugation, the supernatant was added onto the anti-histonecoated microplate, which was incubated for 90 min at RT then washed. Conjugation solution was added,incubated for90 min, and washed. The color was developed by addition of ABTS (2,2 ′-azino-di-[3-ethylbenzthiazoline sulfonate]) substrate solution (1 mg/ml). After shaking incubation for20 min the absorbance was measured at 405 nm.
Insulin secretion
At the end of the different treatments,cells were washed and incubated for 2 h with KRB solution containing 2.8 mmol/L glucose (basal) followed by incubation with medium containing 16.7 mmol/L glucose (stimulatory). All incubation media were kept in −20 °C until insulin secretion was assessed.Insulin was quantified using the rat insulin ELISA kit (DRG diagnostics, Germany, cat. no. EIA-2048).
Statistical analysis
Data were presented as the statistical mean ±SEM (standard error of mean) of N =4 independent experiments. In measurements of some parameters, where N =3 was mentioned, samples from only 3 experiments were processed.For experiments including Ins1E cells,used cells were from different passages ranging from 30 to 50. Statistical analyses between different groups were examined by one-way analysis of variance (ANOVA). Student’s t test was used to compare the difference between 2 groups and as a post hoc test whenever ANOVA was significant.Ap value of statistically significant in all analyses.
Results
FABP3 is involved in FA‑stimulated functions of insulin‑producing tissue
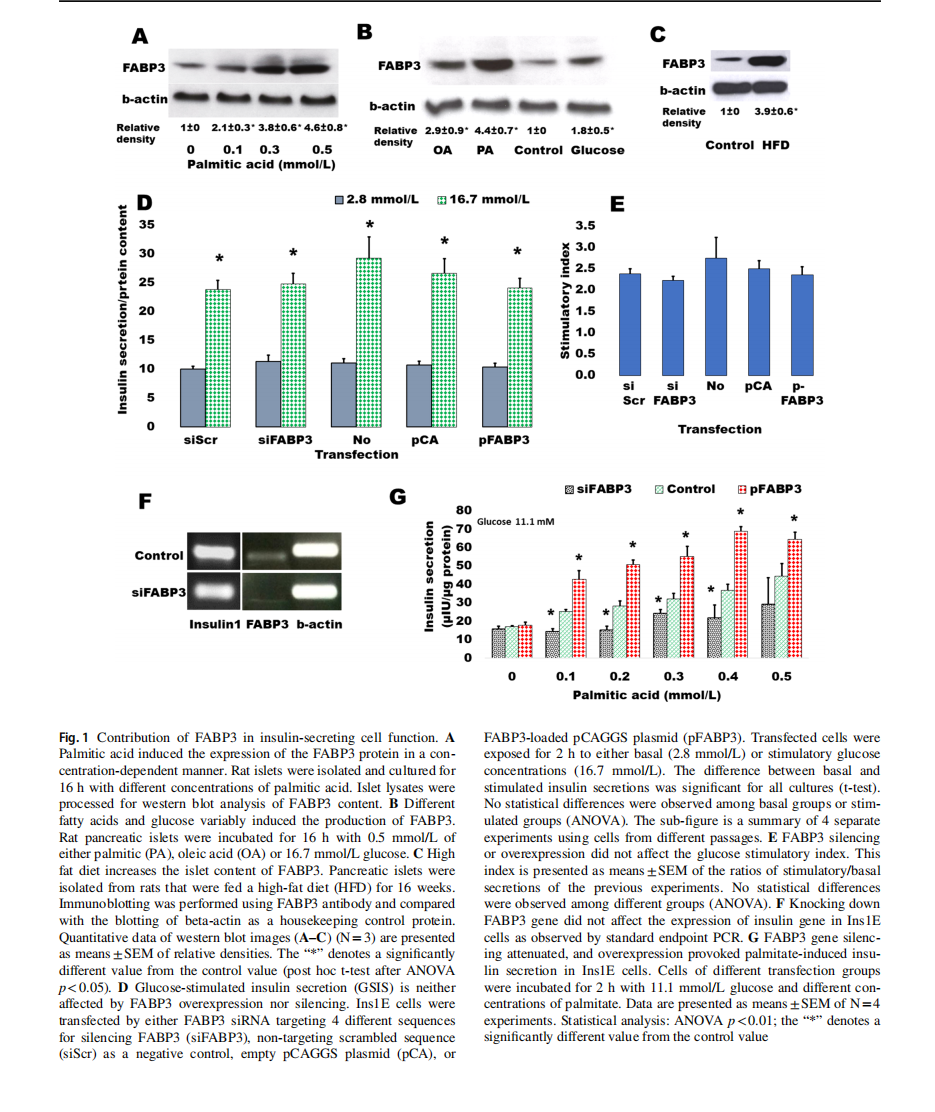
The dependence of FABP3 expression in insulin-secreting cells from FA supply was first investigated (Fig. 1). Rat islets were incubated overnight with ascending concentrations of palmitic acid ranging from 0 (control)to 0.5 mmol/L(Fig. 1A).It was found that FABP3 production significantly increased with palmitic acid concentration. In confirmation with the previous study [8], the production level ofFABP3 protein in response to different stimulators was variable (Fig. 1B). Palmitic acid increased the cellular level ofFABP3 more than oleic acid. Glucose also stimulated the production of FABP3 in islet cells. The results were confirmed in vivo: a significantly higher level of FABP3 was found by western blotting in protein lysates of pancreatic islets isolated from rats that were fed on a high-fat diet for16 weeks (Fig. 1C).
To investigate the role of FABP3 in the glucose-stimulated insulin secretion (GSIS) process, we exposed Ins1E cells, in which FABP3 gene was down-knocked or overexpressed, to low (2.8 mmol/L) and high (16.7 mmol/L) glucose. The results revealed that glucose could significantly stimulate insulin secretion in cells of all treatments(Fig. 1D). The stimulatory index (Fig. 1E), which reflects the elevation of stimulated secretion over the basal secretion, was also similar in different cell treatments. Furthermore, the expression level of the insulin1 gene did not change in cells, where FABP3 was knocked down (Fig. 1F).
The effect of FABP3 on FA-stimulated insulin secretion was examined (Fig. 1G) by exposure of Fabp3-down knocked and overexpressing Ins1E cells to ascending palmitate concentrations in the presence of a constant medium glucose concentration (11.1 mmol/L). FABP3 gene overexpression significantly enhanced the stimulatory effect of palmitic acid,whereas its silencing significantly reduces this stimulatory effect. In most palmitate concentrations, this stimulatory effect was blunted since no significant difference was observed in palmitate-stimulated secretion when compared with the value in the absence of palmitate.
Taken together, these results indicate that FABP3 plays a role in GSIS only in the presence of fatty acids. Also, the insulin stimulatory effect of FAs can be blunted by the inhibition of FABP3 .
FABP3 silencing inhibits FA uptake into insulin‑secreting cells
Ins1E cells were transfected by siRNA to knock down FABP3 . These cells were incubated with fluorescent fatty acid and the kinetics of the intracellular up-taken fluorescence was read by fluorescence plate reader (Fig. 2A). Silencing of the FABP3 (siFABP3) gene reduced fatty acid uptake, compared with the corresponding siScr control uptake. The blank result shown in the figure shows the background fluorescence of cell-free wells.The endpoint FA uptake of the same cells after 1h of incubation with fluorescent lauric acid was significantly reduced by silencing of the FABP3 gene (Fig. 1B).
FAs are transported from the extracellular to the intracellular compartments by another protein: the cell membrane-bound fatty acid translocase CD36 [30]. The observation of decreased FA uptake by silencing FABP3 led us to explore the expression pattern of CD36 in these cells. Silencing FABP3 downregulated, while FABP3 overexpression upregulated CD36 expression, respectively
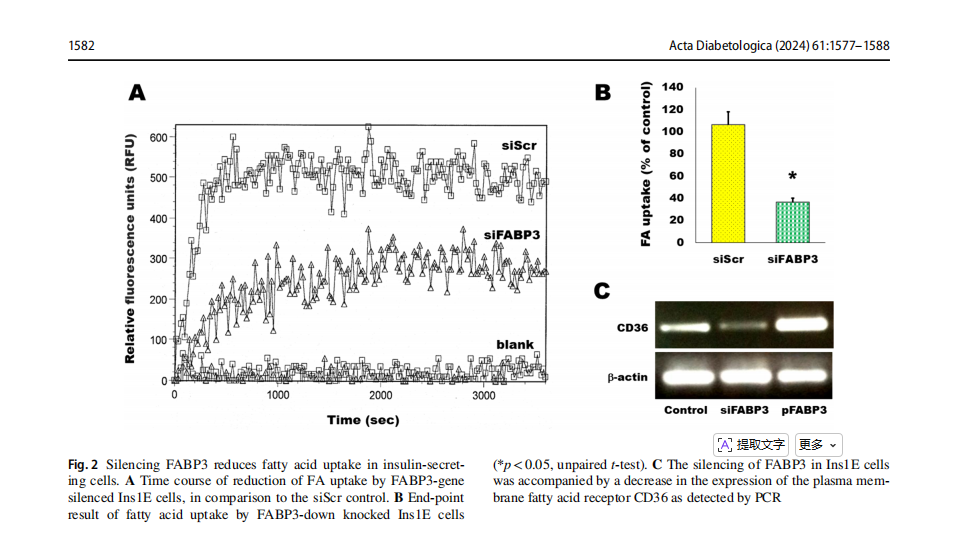
(Fig. 2C). Therefore, it is not anticipated that CD36 intracellularly transfers extra fatty acids that are unable to be processed following FABP3 silencing. Thus, both FA transport across cell membrane and cellular uptake are decreased by Fabp3 silencing in insulin-producing cells.
Lipid accumulation in insulin‑producing cells is inhibited by knocking down FABP3
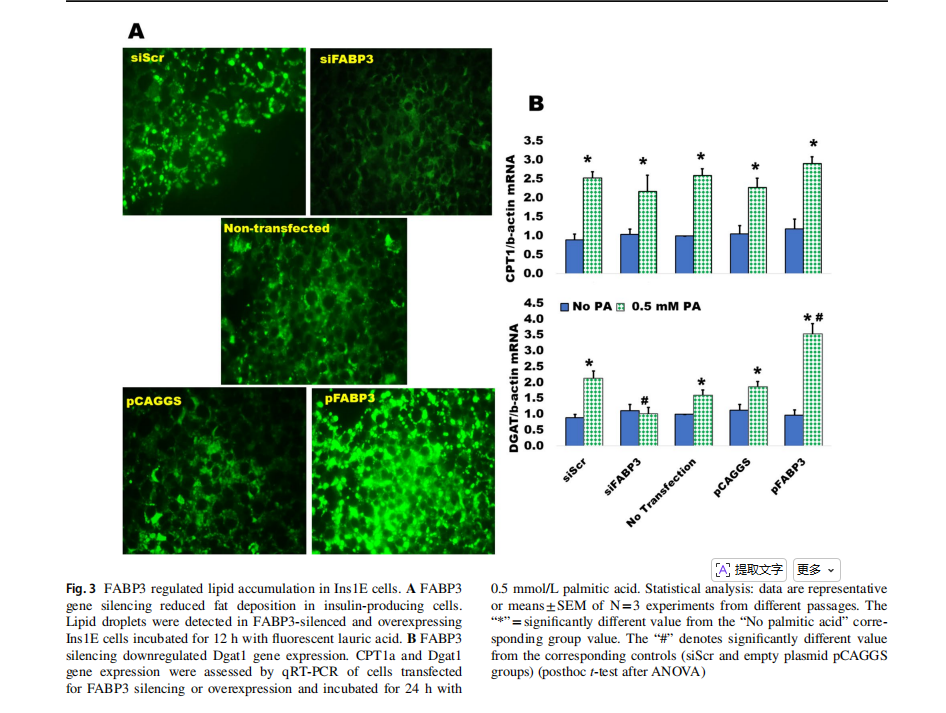
The silencing of FABP3 gene inhibited lipid accumulation in Ins1E insulin-secreting cells, as compared to the siScr control, while FABP3 gene overexpression increased the accumulation of lipid droplets (Fig. 3A).
Fatty acid oxidation and triglyceride formation (lipid accumulation) are regulated by carnitine palmitoyltransferase 1a (CPT1a) and diacylglycerol acyltransferase 1(Dgat1), respectively [8]. Both are upregulated by the influx of fatty acids. The expression of both genes was determined in FABP3-silenced and overexpressing Ins1E cells incubated with palmitic acid. Only Dgat1 expression was blunted in palmitate-stimulated FABP3-silenced cells(Fig. 3B). This effect was reversed in FABP3-overexpressing cells, indicating that FABP3 level controls Dagt1 expression and TG accumulation. FABP3 silencing did not affect the palmitate-induced CPT1a expression level, when compared to the siScr control value. This effect was similar in FABP3-overexpressing cells.
FABP3 silencing inhibits FA‑induced inflammatory response and apoptosis in insulin‑producing cells
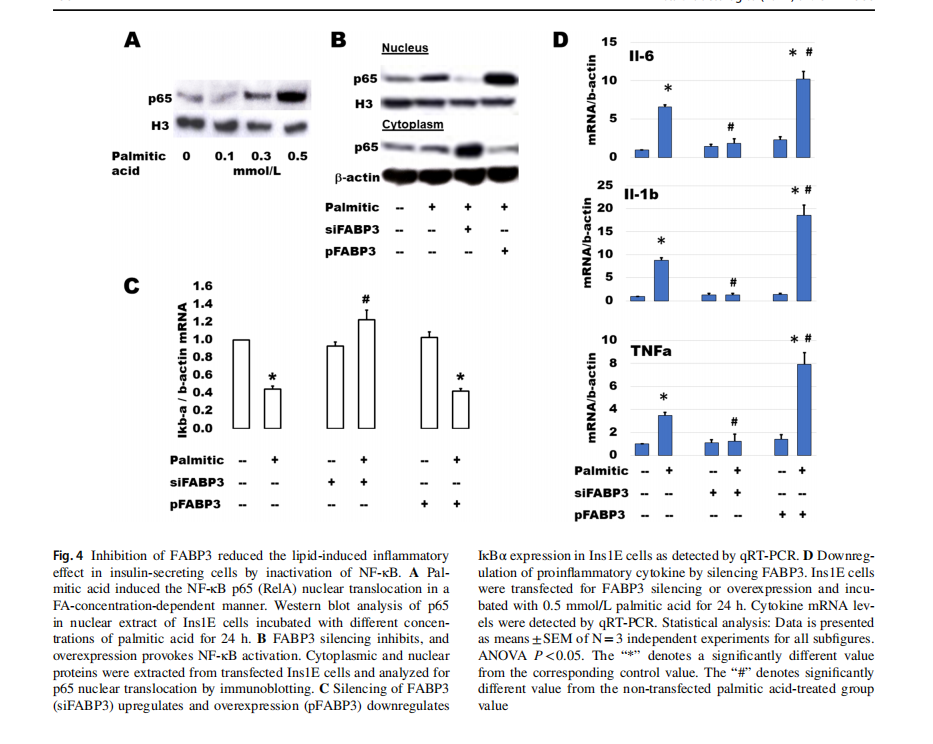
Palmitic acid stimulates β-cell expression of main cytokines suspected of inducing β-cell dysfunction [31]. NF-κB plays a main role in palmitic acid-stimulated inflammation and β-cell failure [32]. The activation of NF-κB includes the translocation of p65 (RelA) protein from the cytosolic into the nuclear compartments via downregulation of IkBα. After incubation of Ins1E cells with different concentrations of palmitic acid for 24 h, p65 increased gradually with the increment of palmitic acid concentration (Fig. 4A). To study the effect of FABP3 silencing on the FA-induced inflammatory response, Ins1E cells were transfected for FABP3 silencing or overexpression and incubated with palmitic acid for 24 h. Silencing of FABP3 inhibited the p65 protein nuclear translocation. The reverse action was observed for FABP3 overexpression (Fig. 4B). Palmitic acid downregulated the expression of IkBα in control and FABP3-overexpressing cells, while FABP3 silencing hindered this downregulation (Fig. 4C). Taken together, NF-κB was not activated in FABP3 -down knocked cells. Similar results were obtained for the expression of the proinflammatory cytokines Il-6, Il-1β, and TNFα (Fig. 1D). Incubation of Ins1E cells with palmitic acid upregulated the expression of these cytokines. This effect is increased in FABP3-overexpressing cells. In contrast, the expression of these inflammatory cytokines
was not affected by palmitic acid in FABP3-silenced cells, and the values were significantly lower than that in the corresponding control (siScr-transfected cells).
We have further analyzed some apoptosis parameters in the same samples (Fig. 5). It is well-known that islets cultured in the presence of high levels of FAs exhibit the characteristic events of apoptosis including DNA fragmentation, increased caspase activity, ceramide formation, and expression of apoptotic genes [33]. The present findings demonstrate that this palmitate-induced effect on DNA fragmentation (Fig. 5A) and caspase 3 activation (Fig. 5B) significantly increased in FABP3-overexpressing Ins1E cells. In contrast, the FABP3-knocked down cells did not respond with apoptosis to exposure to palmitic acid: no changes were noticed regarding DNA fragmentation and cleaved caspase 3 in these cells. Cell death increased indirectly proportional relationship with palmitic acid concentration (Fig. 5C). This could be inhibited by FABP3 silencing.
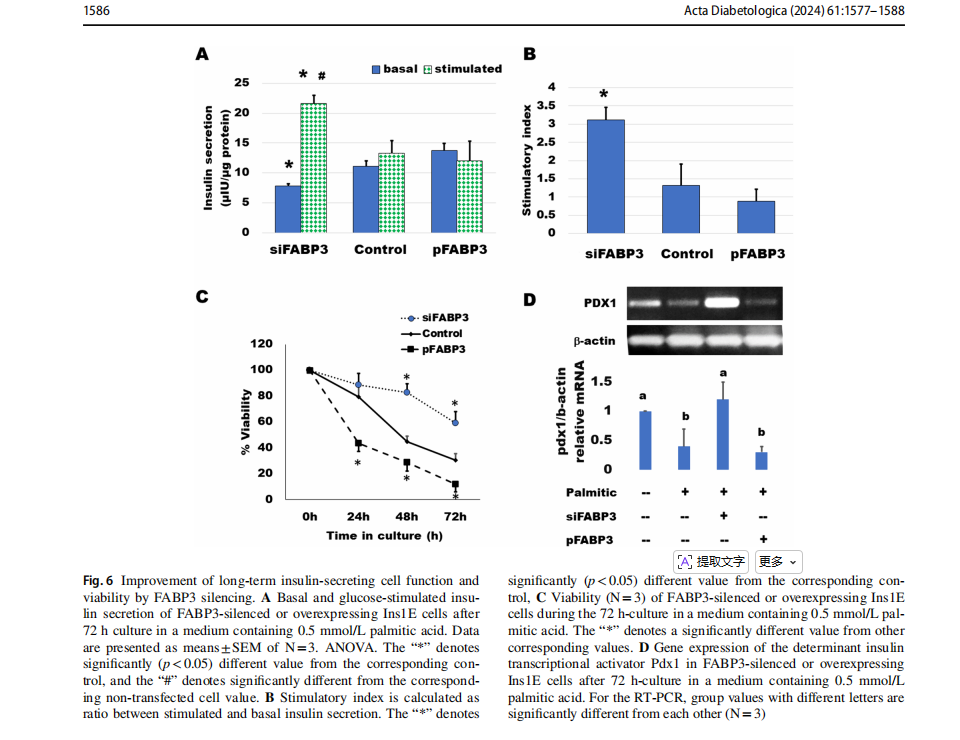
Knocking down FABP3 maintained long‑term insulin‑secreting cell function
Cells were transfected for silencing or overexpression ofFABP3 and incubated to attach for 4 h. Then, the medium was replaced by that containing palmitate and incubated for 3 days. GSIS was found to be diminished by culture with 0.5 mmol/L palmitic acid (Fig. 6). Most cells looked dead and unattached. Glucose could not stimulate insulin secretion (Fig. 6A). The situation was similar in cultures ofFABP3 overexpressing cells. However, in FABP3-silenced cells, the basal insulin secretion was significantly lower than that of both non-transfected and FABP3-overexpressing cells. The glucose-stimulated insulin secretion was significantly higher than its basal value and the value of non-transfected and overexpressing cells. The stimulatory index (Fig. 6B) was significantly higher in these FABP3 -silenced cells than that of other treatments. The cell viability over the 3-day culture with palmitic acid (Fig. 6C) was
maintained by knocking down FABP3 in comparison to nontransfected and FABP3-overexpressing cells.The functionalβ-cell-specific transcription factor PDX1 gene expression confirmed the results of insulin-secreting function in the same cultures (Fig. 6D). Knocking down FABP3 was found to significantly preserve PDX1 gene expression, while its expression decreased in other cultures treated with palmitic acid, as determined by qRT-PCR.
Discussion
Lipotoxicity is a recognized mechanism of the loss of β-cell function in the pathophysiology of type 2 diabetes [34]. When entering β-cell,FAs mostly bind to fatty acid binding protein 3 to be metabolized. The main hypothesis in this work was: knocking down FABP3 in beta cells will reduce FA uptake and consequently the islet lipotoxicity and prevent
islet cell apoptosis.Furthermore,inhibition of the fatty acid uptake by ß-cells might blunt the FA-induced inflammatory outcome [35].This might contribute to improvement of beta cell survival and function, even in high fat conditions, and by that prevent or at least delay the incidence of diabetes.
The present study demonstrated that neither FABP3 inhibition nor overproduction affected the GSIS in the absence ofFAs in culture medium.In contrast,in presence of stimulatory glucose concentration, there was a promotion in FAinduced insulin secretion from cells overexpressing FABP3, and a reduction of secretion from cells with down-knocked FABP3 . We used a seemly high glucose concentration in this experiment because it is known that in beta cells lipotoxic effects are induced only in the presence of elevated glucose concentrations, since glucose is the determinant player in intracellular fatty acid destination [36, 37]. This finding minimizes the role of FABP3 in the absence ofFAs and supports its role in the process of FA induction of
insulin secretion. It also confirms the previous reports that, in contrast to glucose, exposure to FAs does not induce key changes in glucose-related gene expression in Ins1E cells and human islets [38]. This finding also indicates that inhibition ofFABP3 may not be hazardous to beta cell glucosestimulatory function.
The silencing ofFABP3 gene in beta cells decreased both fatty acid uptake and the consequent fatty acid accumulation in insulin-secreting beta cells as proven by immunofluorescence. Also, it did not affect the expression of fatty acid transport and oxidation gene Cpt1, while it downregulated Dgat1 and lipid accumulation. Defective fatty acid uptake was reported in several models of FABPs deficiency [7,39–42]. A positive correlation exists between FABP levels and FA uptake [7]. In agreement with the decreased TG accumulation (lipid droplets) in Fabp3-knocked down cells,it was reported that in FABP3 KO mice, deficiency ofFABP3 prevented the increase in cellular TG levels in skeletal muscle and heart that occur after long-term HFD [43].
Increasing the lipid availability and accumulation in beta cells was reported to induce many changes. It influences membrane and organelle structures, lowers membrane tension, and alters physicochemical properties of β-cells [44]; it significantly increases cellular ceramide levels that have been linked to pancreatic beta cell apoptosis and dysfunction[45]; and it induces apoptosis by stimulating the exhaustion of endoplasmic reticulum calcium stores,leading to the activation of inflammation and apoptotic pathway [46]. In the present study, FABP3 gene silencing caused a reduction of the FA availability and lipid accumulation inside
the beta cells. As a result, our data revealed a reduction in inflammation and apoptosis in beta cells in response to fatty acids. The reduction of inflammation included lowering the expression of inflammatory cytokines, the inactivation of NF-κB, and the prevention of the deactivation of IkBa. The reduction of apoptosis included the inactivation of caspase3 and prevention of DNA fragmentation. The mechanisms of beta cell inflammation and apoptosis include the activation of inflammatory signals like NF-κB, IL1b, IL-6, and TNFα, which impair insulin secretion. IL-1b activates the NF-κB signaling leading to dysregulation ofFA metabolism and DNA fragmentation, resulting in beta cell dysfunction[47, 48]. The present results agree with these mechanisms and show that silencing of Fabp3 can minimize the lipid accumulation in beta cells and prevent the causative factors of inflammation and apoptosis.In other cell systems, and in agreement with our results, FABP3 deficiency also downregulated the expression of cleaved caspase 3 and Bax and upregulated the level of Bcl-2 in myocytes and exerted protective effects against ischemic heart injuries by decreasing cardiac myocyte apoptosis [13]. The present results agree with this protective role of FABP3 deficiency against beta cell apoptosis and dysfunction.
This protective role of FABP3 silencing was confirmed by a 72-culture with palmitate, and the data revealed normal Ins1E beta cells exhibiting normal viability,gene expression pattern, and insulin secretory function, despite the exposure to palmitic acid. The present data reveals that inhibition ofFABP3 may protect pancreatic beta cells from lipotoxicity. In this context, we have reviewed some antidiabetic plants
and found some natural products that can compete with FAs to bind FABP3 [20]. These competitive inhibitors might be safe to apply,but this remains to be elucidated. Other chemically designed commercial inhibitors are also available [49]
Conclusion
Fatty acid binding protein 3 is produced in pancreatic beta cells to bind FAs and allow for FA metabolism. Longterm exposure of these cells to FA leads to inflammatory response, apoptosis, and dysfunction. In the present study, we have manipulated the expression of FABP3 by silencing or overexpression in Ins1E beta cells. Knocking down Fabp3 was found to reduce FA uptake and lipid accumulation in these beta cells. This reduction protected cells from apoptosis and maintained their long-term function. FABP3 inhibitors may be useful to prevent the deleterious effects of FA binding and lipid accumulation in pancreatic islets resulting in ß-cell loss and contributing lastly to the pathogenesis of type 2 diabetes.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00592-024-02325-x.
Funding Open access funding provided by The Science,Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This study is supported by the Egyptian Science,Technology &Innovation Funding Authority (STDF grant ID. BARG 37106).
Declarations
Conflict of interest The authors declare that they have no conflict of interest.
Ethical standard For animal experiments, the“Principles of laboratory animal care” (NIH publication No. 86-23,revised 1985) were fol- lowed, and the study was approved by the institutional review board of Damietta University.
Informed consent For this animal study, informed consent is not required.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence,and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use,you will need to obtain permission directly from the copyright holder.To view a copy of this licence,visit http://creativecommons.org/licenses/by/4.0/.



This article is excerpted from the 《Acta Diabetologica》 by Wound World.

- 星期四, 04 6月 2026
Association between triglyceride‑glucose index and the risk of heart failure hospitalization in older diabetic patients received right ventricular pacing: a retrospective cohort study
Bingqi Fu1 · Yu Yu1 · Sijing Cheng1 · Hao Huang1 · Tianxin Long1 · Juweig Yang1 · Min Gu1 · Chi Cai1 · Xuhua Chen1 · Hongxia Niu1 · Wei Hua
Received: 23 February 2024 / Accepted: 11 June 2024 / Published online: 19 June 2024 © The Author(s) 2024
Abstract
Background The prognostic value of triglyceride-glucose (TyG) index is not yet known for older diabetic patients received right ventricular pacing (RVP). We aimed to investigate the association between TyG index and the risk of heart failure hospitalization (HFH) in older diabetic patients received RVP.
Methods This study was conducted between January 2017 and January 2018 at Fuwai Hospital, Beijing, China, and included older (age≥65 years) diabetic patients that received RVP for the first time. TyG index were obtained before implantation. The primary endpoint was HFH.
Results A total of 231 patients were divided into three groups according to the tertiles of TyG index:<8.5 (T1, N=77), 8.5–9.1 (T2, N=77), and>9.1 (T3, N=77). T3 group had higher rate of HFH (Log-rank=11.7, P=0.003). Multivariate analyses showed that, TyG index served as an independent predictor for HFH, both as numerical variable (HR=1.94, 95% CI 1.21–3.11, P=0.006), and as categorical variable (HR=2.31, 95% CI 1.09–4.89, P=0.03). RCS demonstrated that the risk of HFH was relatively low until TyG index exceeded 8.8, beyond which the risk began to increase rapidly (P-non-linear=0.006).
Conclusion Preimplantation TyG index emerges as a robust, independent predictor for HFH in older diabetic patients received RVP, and TyG index>8.8 might be the optimal cut-off value.
Keywords Triglyceride-glucose index · Older · Diabetes · Right ventricular pacing
Abbreviations
AF Atrial fibrillation
CABG Coronary artery bypass grafting
CKD Chronic kidney disease
CVD Cardiovascular disease
eGFR Estimated glomerular filtration rate
FBG Fasting blood glucose
HbA1C Hemoglobin A1C
HDL-C High-density lipoprotein cholesterol
HEC Hyperinsulinemic euglycemic clamp
HF Heart failure
HFH Heart failure hospitalization
HOMA-IR Homeostasis model assessment of insulin
resistance
IR Insulin resistance
LDL-C Low-density lipoprotein cholesterol
LVEF Left ventricular ejection fraction
NT-proBNP N-terminal pro-brain natriuretic peptide
PCI Percutaneous coronary intervention
PPMI Permanent pacemaker implantation
RCS Restricted cubic splines
RVP Right ventricular pacing
T2DM Type 2 diabetes mellitus
TC Total cholesterol
TyG index Triglyceride-glucose index

- 星期三, 03 6月 2026
Differences in physiological response to an oral glucose tolerance test in adult Maasai men and women
Dirk Lund Christensen1 · Cathrine Olesen Emborg1,2 · Kaushik Laxmidas Ramaiya3 · Venance Philip Maro4 · Ib Christian Bygbjerg1 · Joseph Sironga4,5 · Jens Juul Holst6 · Kajiru Kilonzo4 · Bolette Hartmann6 · Flemming Dela6,7 · Steen Larsen6,8 · Jørn Wulff Helge6
Received: 13 September 2025 / Accepted: 26 April 2026 © The Author(s) 2026
Abstract
Aims Rural, agro-pastoralist Maasai in East Africa exhibit low prevalence of diabetes, yet little is known about their physi-ological response to glucose loads and whether sex has an impact on glucose metabolism, including incretin hormones.
Methods We included 58 (29 men, 29 women) adult Maasai without diabetes living in rural Tanzania. Clinical background characteristics were measured, and they were exposed to an oral glucose tolerance test (OGTT) after an overnight fast. Plasma glucose, insulin/C-peptide, glucagon, glucose-dependent insulinotropic polypeptide (GIP), and glucagon-like pep-tide-1 (GLP-1) were analysed.
Results Mean age was 34.8 (range 17–65) years, and mean body mass index (BMI) was 20.3 (range 14.0-30.9) kg/m2 with two individuals being overweight and 14 being underweight. Men had a higher mean fasting glucose concentration (5.2 vs. 4.9 mmol/L, p=0.031), while women exhibited a higher OGTT-derived mean GLP-1 concentration at 2-h (8.5 vs. 5.1 pmol/L, p=0.005). Total area-under-the-curve (tAUC) for GLP-1 was higher in women compared to men (1336 vs. 870 pmol x min, p=0.011). Sex- and BMI-adjusted regression analyses for tAUC showed higher values of insulin, C-peptide and GLP-1 in women (p<0.10).
Conclusions Sex differences were found in fasting glucose and OGTT-derived insulin/C-peptide and GLP-1 concentrations. Research using more sophisticated methodology is needed to further explain the glucose metabolism phenotype in Maasai
Keywords Oral glucose tolerance test · Incretin hormones · Sex differences · Maasai · Sub-Saharan Africa

- 星期二, 02 6月 2026
Long-term imaging characteristics of the pancreas related to diabetes induced by immune checkpoint inhibitor therapy
Seiji Tomofuji1 · Shin Urai2 · Kei Yoshino2,3 · Hironori Bando1 · Yushi Hirota1
Received: 25 November 2025 / Accepted: 9 March 2026 © The Author(s) 2026
Abstract
Background Immune checkpoint inhibitor (ICI)-induced diabetes is a rare endocrine immune-related adverse event, and long-term pancreatic magnetic resonance imaging (MRI) findings after disease onset have not been well characterized.
Case presentation An individual developed abrupt hyperglycemia during pembrolizumab therapy for recurrent renal cell carcinoma and subsequently required insulin therapy. Serum C-peptide rapidly declined to below the detection threshold, whereas pancreatic enzyme levels remained within normal limits throughout follow-up. MRI performed 1 day after diagno-sis demonstrated diffuse high signal intensity on diffusion-weighted imaging (DWI) and reduced apparent diffusion coeffi-cient (ADC) values throughout the pancreas. Serial imaging over 25 months showed progressive pancreatic atrophy, whereas the DWI/ADC features persisted.
Conclusion This case provides a new descriptive longitudinal radiological observation of persistent diffusion-related pancre-atic MRI findings and progressive pancreatic atrophy after the onset of ICI-induced diabetes.
Keywords Type 1 diabetes · Immune checkpoint inhibitor · PD-1 antibody · Immune-related adverse event · Magnetic resonance imaging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}